Fig. 7.1
Molecular markers of senescence. Pictures showing some examples of senescence molecular markers, including senescence-associated b-galactosidase (SA-bGal) activity; senescence-associated heterochromatin foci (SAHF), visualized by immunofluorescence microscopy with DAPI staining; and two senescence-associated DNA damage markers: 53BP1 and the phosphorylated histone H2A variant gH2AX (p- gH2AX), visualized by immunofluorescence microscopy using specific antibodies
Senescent cells display molecular characteristics of DNA damage (Fig. 7.1). Markers of a DNA damage response localize at telomeres in senescent cells after serial passage (d’Adda di Fagagna et al. 2003; d’Adda di Fagagna 2008; Herbig et al. 2004), which indicates that the DNA damage response can be triggered by telomere shortening. These markers include nuclear foci of phosphorylated histone H2AX, the localization at double-strand break sites of DNA-repair and DNA-damage checkpoint factors, such as 53BP1, MDC1 and NBS1 (d’Adda di Fagagna 2008; Ruiz et al. 2008). Senescent cells also contain activated forms of the DNA-damage checkpoint kinases Chk1 and Chk2. These markers and others suggest that telomere shortening initiates senescence through a DNA damage response. These characteristics also explain why other DNA damage stressors, such as culture shock, potentially initiate senescence without telomere involvement (Hewitt et al. 2013). In parallel, the redox potential poise of some cells changes in response to chemical modifications. This modification results in altered gene expression, enzyme activity, and signaling pathways. Finally, it results in DNA damage (Nelson and von Zglinicki 2013; Passos et al. 2013).
Large protein and lipid modification is another characteristic of senescent cells (Gasparovic et al. 2013; Vistoli et al. 2013). Oxidation, glycation, cross-linking, and other chemical modifications impair the molecular functions of multiple vital components, including DNA, membranes, the extracellular matrix (ECM), enzymes, and structural proteins. Modifications that accumulate faster than they are repaired or recycled will cause progressive deterioration over time.
Nuclear envelope alterations in senescence nuclear structures, such as the nuclear lamina, nucleoli, the nuclear matrix, nuclear bodies (such as promyelocytic leukemia bodies), and nuclear morphology are altered within growth-arrested or senescent cells. It is especially interesting that multinucleation is probably the consequence of the failure of nuclear envelope breakdown (de la Rosa et al. 2013).
Senescence-associated secretory phenotype (SASP). Senescent cells undergo widespread changes in protein expression and secretion, which ultimately develops into the SASP (Campisi 2011; Campisi et al. 2011). Senescent cells upregulate the expression and secretion of several matrix metalloproteinases that comprise a conserved genomic cluster and interleukins that promote the growth of premalignant epithelial cells. A limited number of cell culture and mouse xenograft studies support the idea that senescent cells secrete factors that can disrupt tissue structure, alter tissue function and promote cancer progression (Bavik et al. 2006; Krtolica et al. 2001; Parrinello et al. 2005). Recent studies on the SASP of human and mouse fibroblasts show it is conserved across cell types and species; moreover, specific secreted factors are strong candidates for stimulating malignant phenotypes in neighboring cells (Coppe et al. 2008, 2010a, b). The idea that a biological process, such as cellular senescence, can be beneficial (tumor suppressive) and deleterious (pro-tumorigenic) is consistent with a major evolutionary theory of aging termed antagonistic pleiotropy (Coppe et al. 2010a). The SASP is possibly the major reason for the deleterious side of the senescence response (Davalos et al. 2010; Rodier 2013).
Consistent with a role in aging, senescent cells accumulate with age in many rodent and human tissues (Campisi 2005). Moreover, they are found at sites of age-related pathology, including degenerative disorders such as osteoarthritis and atherosclerosis (Campisi 2005) and hyperproliferative lesions such as benign prostatic hyperplasia (Castro et al. 2003) and melanocytic naevi (Michaloglou et al. 2008). A limited number of cell culture and mouse xenograft studies support the idea that senescent cells secrete factors that can disrupt tissue structure and function and promote cancer progression (Bavik et al. 2006; Krtolica et al. 2001; Parrinello et al. 2005). Recent studies on the senescence-associated secretory phenotype (SASP) of human and mouse fibroblasts show it is conserved across cell types and species and that specific secreted factors are strong candidates for stimulating malignant phenotypes in neighboring cells (Coppe et al. 2008, 2010a, b).
The idea that a biological process such as cellular senescence can be both beneficial (tumor suppressive) and deleterious (pro-tumorigenic) is consistent with a major evolutionary theory of aging termed antagonistic pleiotropy (Coppe et al. 2010a). The SASP may be the major reason for the deleterious side of the senescence response (Davalos et al. 2010).
As mentioned, in addition to telomere dysfunction, cellular senescence can be elicited by other types of stress, including oncogene activation (Collado and Serrano 2006). This phenomenon is not observed for oncogenic RAS exclusively; many – but not all – of its effectors, including activated mutants of RAF, MEK and BRAF, were shown to cause senescence as well (Braig and Schmitt 2006; Carnero et al. 2003; Courtois-Cox et al. 2008; Chandeck and Mooi 2010). Some oncogenes, such as RAS, CDC6, cyclin E, and STAT5, trigger a DNA damage response (DDR), associated with DNA hyper-replication that appears to be causally involved in oncogene-induced senescence (OIS) in vitro (Bartek et al. 2007; Di Micco et al. 2006; Kenyon and Gerson 2007; Ruzankina et al. 2008). During most of the last decade, OIS has been studied predominantly in cell culture systems, triggering a long debate as to whether or not OIS corresponds to a physiologically relevant phenomenon in vivo. In favour of OIS representing an in vitro phenomenon only is that artificial conditions, such as the use of bovine serum and plastic dishes, as well as the presence of supraphysiologic O2, generate a stress signal that at the very least contributes to triggering a cellular senescence response (Parrinello et al. 2003; Passos and Von Zglinicki 2006). However, conversely, senescence bypass screens have identified several genuine human oncogenes, including TBX2, BCL6, KLF4, hDRIL, BRF1, PPP1CA and others (Vergel and Carnero 2010). Furthermore, virtually all human cancers lack functional p53/pRB pathways, two key senescence- signalling routes (Malumbres and Carnero 2003), and often carry mutations in sets of genes, which are known to collaborate in vitro in bypassing the senescence response.
7.2 Effector Pathways
Cellular senescence pathways are believed to have multiple layers of regulation, with additional redundancy built into these layers (Smith and Pereira-Smith 1996). On the basis of the complementation studies, there are at least four senescence pathways (Duncan et al. 1993). This indicates that in any one immortal cell line, there are probably multiple senescence genes/pathways that are abrogated (Barrett et al. 1994). Many of the functional studies, where a putative senescence gene is overexpressed in cells, indicate that although multiple genes/pathways may be abrogated in a particular cell line, as little as one gene/pathway is required for repair and subsequent reversion to senescence, indicating that senescence is essentially a dominant phenomena (Fig. 7.2).
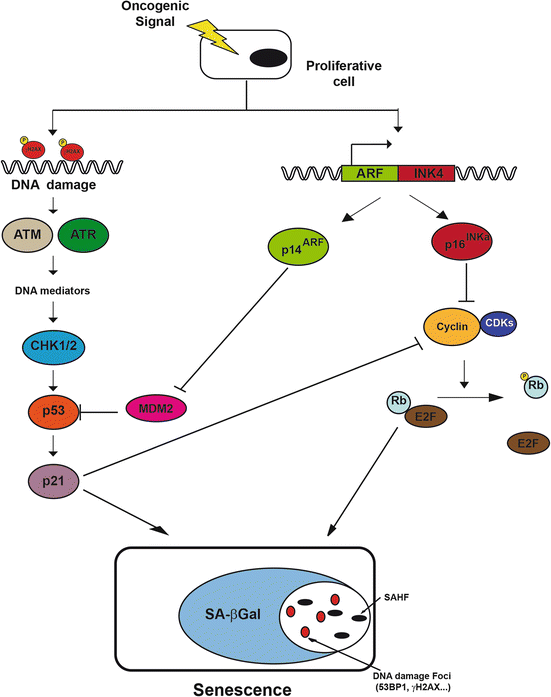
Fig. 7.2
Molecular pathways of oncogenic-induced senescence. When a proliferative cell suffers oncogenic stress, two different pathways leading to cellular senescence are activated: one p53-dependent, induced by DNA damage checkpoint activation, and other p16-Rb-dependent, which stops cell cycle progression. Both pathways interact functionally giving some redundancy to the effector signal
Pathways known to regulate cellular senescence/immortalisation, including the p16INK4a/pRB pathway, the p19ARF/p53/p21CIP1/WAF1 pathway and the PTEN/p27KIP1 pathway, are reviewed in (Carnero and Lleonart 2010; Mooi and Peeper 2006; Schmitt 2007; Serrano and Blasco 2001; Vergel and Carnero 2010). Other genes that have been shown to contribute to a senescence-like phenotype include PPP1A (Castro et al. 2008b), SAHH (Leal et al. 2008a: ME et al. 2006), Csn2, Arase and BRF1 (Leal et al. 2008b), PGM (Kondoh et al. 2005), IGFBP3 and IGFBPrP1 (Fridman et al. 2007), PAI-1 (Kortlever and Bernards 2006; Kortlever et al. 2006), MKK3 (Wang et al. 2002), MKK6 (Haq et al. 2002; Wang et al. 2002),Smurf2 (Zhang and Cohen 2004) and HIC-5 (Shibanuma et al. 1997). All these genes have shown to be related to human tumorigenesis. However, all these genes and their pathways, as indicated earlier, can act in sequential steps conforming a well-regulated process.
The dynamics of senescence exhibit two different steps: cell cycle arrest and further acquisition of senescence features, which includes permanent arrest, termed geroconversion.
Senescence effector pathways converge at the point of cell cycle arrest through CDK inhibition. Therefore, most pathways known to be involved in senescent arrest impinge either directly or indirectly on this process. Namely, the most known effector pathways are the p16INK4a/pRB pathway, the p19ARF/p53/p21CIP1 pathway and the PI3K/mTOR/FoxO pathway (Carnero and Lleonart 2010; Mooi and Peeper 2006; Schmitt 2007; Serrano and Blasco 2001; Vergel and Carnero 2010), all of which exhibit a high degree of interconnection. Two pathways have been proposed to be responsible for the acquisition of irreversible arrest and geroconversion: the pRB pathway and the mTOR pathway. If geroconversion is not activated, cells are only transiently arrested with the possibility of resuming growth once the proliferation constraints have been eliminated (Ferbeyre et al. 2002; Ruiz et al. 2008). It has also been shown that if mTOR is activated under conditions of proliferative arrest, then arrest becomes permanent and the cell undergoes senescence (Blagosklonny 2010; Demidenko and Blagosklonny 2008). This can also be accomplished by producing permanent changes in the chromatin, especially at E2F transcription sites, which results in a blockade of transcription of proliferative genes (Narita et al. 2003). It has been shown that permanent inactivation of pRb, perhaps in combination with phosphatases (Castro et al. 2008a), may signal for the differential recruitment of silencers to the heterochromatin of promoter sites. Human cells show heterochromatin compaction during senescence (SAHF for senescence-associated heterochromatin foci), which is dependent on the pRb pathway (Narita and Lowe 2004). These SAHFs cause stable silencing of cell cycle genes and seem to be a factor in the stability of permanent arrest during senescence. Also, the role of senescence in embryonic development seems to be dependent on the pRb pathway through CDK inhibitors p21CIP1 and p15INK4b but independent of other cell cycle inhibitors, DNA damage or p53. This senescence during embryonic development seems also regulated by the PI3K/FOXO and TGFb/SMAD pathways (Munoz-Espin et al. 2013; Storer et al. 2013).
The absence of p53 function induced by dominant negative mutants, specific p53 antisense, oligonucleotides or viral oncoproteins (such as SV40 T antigen or HPV16 E6) is sufficient to substantially extend the lifespan of several cell types in culture (Wynford-Thomas 1996). Consistent with this, senescence is associated with a switch-on of the transactivation function of p53 in culture (Bond et al. 1996). Coincident with telomere shortening, DNA-damage checkpoint activation and associated genomic instability, p53 is also activated in vivo (Chin et al. 1999). Deletion of p53 attenuated the cellular and organismal effects of telomere dysfunction, establishing a key role for p53 in the shortening response (Chin et al. 1999). Other p53 regulatory proteins are also involved in senescence. MDM2 protein has p53 ubiquitin ligase activity and forms an autoregulatory loop with p53 (Ashcroft et al. 2000). Overexpression of MDM2 targets p53 for degradation and induces functional-p53 loss (Blaydes and Wynford-Thomas 1998). The product of another gene up-regulated in senescence – p14ARF – can release p53 from inhibition by MDM2 and cause growth arrest in young fibroblasts (Blaydes and Wynford-Thomas 1998). Seeding mouse embryonic fibroblasts (MEFs) into culture induces the synthesis of ARF protein, which continues to accumulate until the cells enter senescence (Kamijo et al. 1997). MEFs derived from ARF-disrupted mice (Kamijo et al. 1997) or wild type fibroblasts expressing an efficient ARF antisense construct (Carnero et al. 2000b) are also efficiently immortalised. Concomitant with this observation, overexpression of MDM2 in naïve MEFs produces efficient immortalisation (Carnero et al. 2000b). Activation of p53 induces the up-regulation of the cyclin-dependent kinase (CDK) inhibitor p21WAF1, which has a direct inhibitory action on the cell-cycle machinery (Malumbres and Carnero 2003) and correlates well with the declining growth rate in senescing cultures. In mouse embryo fibroblasts however, the absence of p21WAF1 does not overcome senescence (Carnero and Beach 2004; Pantoja and Serrano 1999). This suggests that at least one additional downstream effector is needed for p53-induced growth arrest in senescence. In contrast, a different behaviour is observed in human cells, where elimination of p21 by a double round of homologous recombination is sufficient to bypass senescence (Brown et al. 1997). Other p53 effectors might be also involved, such as 14-3-3 and GADD45, which inhibit G2/M transition, or downregulation of Myc (Ho et al. 2005).
The retinoblastoma tumor suppressor pathway, pRb, has also been related to senescence. Overexpression of pRb, as well as some of the regulators of the pRb pathway such as CDK inhibitors, leads to growth arrest mimicking the senescent phenotype (Carnero et al. 2003). Moreover, inactivation of pRb by viral oncoproteins such as E7, SV40 large T antigen and E1A leads to extension of lifespan (Haferkamp et al. 2009; Jarrard et al. 1999; Ye et al. 2007). Other members of the pocket protein family comprising pRb, p130 and p107 may also be involved. In MEFs, p130 levels decrease with population doublings and MEFs from triple pRb, p130 and p107 knockout mice are immortal (Mulligan and Jacks 1998). Nevertheless, since a certain degree of complementation has been observed among the pocket protein family (Mulligan and Jacks 1998), it is difficult to assess the role of each protein in replicative senescence.
Given that p16INK4a functions to inhibit the inactivation of pRb by CDKs (Carnero and Hannon 1998), a loss of functional p16INK4a may be expected to have similar consequences with the loss of functional pRb. Several types of human cells accumulate p16INK4a protein as they approach senescence (Palmero et al. 1997). Senescent fibroblasts may contain p16INK4a levels at least 40-fold greater than early passage cells. The deletion of p16INK4a is common in immortalised tumor cell lines (Okamoto et al. 1994), and several non-tumorigenic in vitro immortalised cell lines also lack functional p16INK4a protein. Expression of p16INK4a-specific antisense in naïve MEFs increases the probability of immortalisation of these cells (Carnero et al. 2000b). In accordance with this observation, mice cells which are made nullizygous for p16INK4a by targeted deletion undergo immortalisation more readily than normal control cells (Krimpenfort et al. 2001; Sharpless et al. 2001), although they show normal senescence kinetics. Knockout mice for p16INK4a proteins develop normally to adulthood and are fertile, indicating that the individual INK4 proteins are not essential for development. p16INK4a deficiency, however, results in a low susceptibility to spontaneous tumor development and increased tumor susceptibility under specific carcinogenic protocols (Krimpenfort et al. 2001; Sharpless et al. 2001). A cross-talk among the different pathways involved in senescence has been found. This cross-talk might ensure the correct functioning of the senescence program. Moreover, genes such as myc that are involved in all the pathways are able to bypass senescence in human primary cells. Myc can bypass CDK4/6 inhibition by activating CDK2-cyclinA/E complexes and inducing the Cdk-activating phosphatase Cdc25A (Amati et al. 1998). Moreover, myc induces degradation of p27, thus influencing the inhibitory effects of PTEN. Finally, expression of myc induces telomerase activity by activating the transcription of the catalytic subunit (Wang et al. 1998a). The overall result is a single step immortalisation of human cells induced by myc gene amplification (Gil et al. 2005).
PI3K/AKT/mTOR/FoxO constitutes an important pathway regulating the signaling cascades of multiple essential biological processes (Blanco-Aparicio et al. 2007; Carnero 2010; Lacal and Carnero 1994). Many components of this pathway are genetically altered in cancer cells. AKT is a master kinase that phosphorylates MDM2 (among other proteins) and promotes its translocation to the nucleus, where it negatively regulates p53 function (Gottlieb et al. 2002). One of the most conserved functions of AKT is its role in cell mass increase through the activation of the mTOR complex 1 (mTORC1 or the mTOR/raptor complex), which is regulated by both nutrients and growth factor signaling. mTORC1 is a critical regulator of translation initiation and ribosome biogenesis and plays an evolutionarily conserved role in cell growth control (Sengupta et al. 2010). PI3K has been related to the induction of cellular senescence in several ways that are still not fully understood. Early works from Collado and colleagues, (Collado et al. 2000) suggest that PI3K inhibition induces senescence through the activation of p27kip1. However, further works also indicated that the overexpression of active P110a (catalytic subunit of PI3K) or AKT induces oncogene-induced senescence in primary cells in culture and in vivo (Carnero and Beach 2004; Di Cristofano et al. 2001; Lorenzini et al. 2002; Renner and Carnero 2009; Trotman et al. 2006). On the other hand, loss of PTEN triggers cellular senescence through a p53-dependent mechanism (Chen et al. 2005) and results in indolent prostate cancer. Therefore, concomitant or sequential loss of PTEN and p53 results in a dramatic acceleration of prostate tumorigenesis. Studies in murine mouse models have shown that p53 is the preferred mutation upon PTEN loss. In constitutively active AKT or PI3K transgenic models, an increase in benign lesions are observed if senescence is induced upon AKT activation (Blanco-Aparicio et al. 2010; Renner et al. 2008).
AKT activation can also stimulate proliferation through multiple downstream targets and impinge on cell-cycle regulation. AKT phosphorylates some members of the FoxO family while they are present in the nucleus, thus creating binding sites for 14-3-3-sigma proteins that trigger their export from the nucleus. Through this mechanism, AKT blocks the FoxO-mediated transcription of target genes that promote apoptosis, cell-cycle arrest, and metabolic processes (Calnan and Brunet 2008; Zanella et al. 2010).
FoxO transcription factors are an evolutionary conserved subfamily that regulate a number of cellular processes involved in cell-fate decisions in a cell-type- and environment-specific manner, including metabolism, differentiation, apoptosis and proliferation (Greer and Brunet 2008). A key mechanism by which FoxO determines cell fate is through regulation of the cell cycle machinery. FoxO plays a crucial role in regulating cellular senescence by controlling the expression of a number of cell cycle regulators, including p27kip1 (Collado et al. 2000). Moreover, overexpression of FoxO or p27KIP1 in primary mouse embryo fibroblasts can recapitulate this phenotype, promoting premature cell cycle arrest, changes in cell morphology and increases in senescence-associated markers. The ability of FoxO to induce G0/G1 arrest is lessened in p27Kip1 and p130 double deficient fibroblasts (Chen et al. 2006), suggesting that both p27Kip1 and p130 are important for mediating FoxO-dependent cellular senescence associated G0/G1 arrest. Further evidence of a role for FoxO in cellular senescence is supported by a recent in vivo study demonstrating that oncogene-induced senescence also involves the repression of the PI3K–PKB signaling pathway and the induction of FoxO (Kyoung Kim et al. 2005).
mTOR is an essential convergence point for the PI3K/AKT/FoxO pathways (Martinez-Gac et al. 2004). mTOR is the master regulator of protein synthesis (Wullschleger et al. 2006). It has been proposed that for growth arrest to become permanent (i.e., undergo senescence), a high level of mTOR activation is necessary (Blagosklonny 2008, 2009). In fact, rapamycin treatment, which inhibits mTOR, can divert senescence into quiescence, allowing the cell to resume growth once conditions are more favorable (Anisimov et al. 2010; Korotchkina et al. 2010). It has been proposed that this contribution is due to the function of mTOR as a sensor of cellular nutrients and energy status as well as growth factor signals. mTOR then integrates those signals and “decides” whether the amount of metabolites and energy are sufficient to permit protein synthesis (Sengupta et al. 2010; Young and Narita 2013).
Crosstalk among the different pathways involved in senescence has been found (Carnero et al. 2000b). This crosstalk might ensure the accurate execution of the senescence program. Moreover, genes, such as myc, that are involved in these pathways bypass senescence in human primary cells. Myc can bypass CDK4/6 inhibition by activating CDK2-cyclinA/E complexes and inducing the Cdk-activating phosphatase Cdc25A (Amati et al. 1998). Moreover, myc induces degradation of p27, which influences the inhibitory effects of PTEN. Finally, expression of myc induces telomerase activity by activating the transcription of the catalytic subunit (Wang et al. 1998a). The overall result is a single step immortalization of human cells induced by myc gene amplification (Gil et al. 2005).
MicroRNAs (miRNAs) are small non-coding endogenous RNA molecules that regulate gene expression and protein coding by base pairing with the 3′ unstranslated region (UTR) of target mRNAs. MiRNA expression is associated with cancer pathogenesis because miRNAs are intimately linked to cancer development. Senescence blocks cell proliferation and represents an important barrier that cells must bypass to reach malignancy. Importantly, certain miRNAs have an important role during cellular senescence, which is also involved in human tumorigenesis (Feliciano et al. 2011).
Several miRNAs are differentially expressed in senescent cells when compared to primary cells, which implies a role for miRNAs in senescence. Recently, miR-34a overexpression has been reported during senescence and can cause senescence in a p53-independent manner through repression of c-myc (Christoffersen et al. 2010). MiR-34a is downregulated in pancreatic cancer cells, neuroblastomas, colon cancer cells, and lung cancer cells (Bommer et al. 2007; Tazawa et al. 2007), which suggests a mechanism for immortalization. The expression levels of miR-29 and miR-30 increase during cellular senescence, and these microRNAs directly repress B-Myb in conjunction with Rb-E2F complexes, which results in senescence (Martinez et al. 2011). MiR-29 is downregulated in cell lymphomas (Zhao et al. 2010), and the overexpression of miR-29 is suppressed during tumorigenicity in lung cancer cells (Fabbri et al. 2007). MiR-449a suppresses pRb phosphorylation, which induces senescence (Marasa et al. 2010; Noonan et al. 2009, 2010). A recent study has shown that miR-449a is downregulated in prostate cancer, which indicates that this miRNA regulates cell growth and viability, in part by repressing the expression of HDAC-1 (Noonan et al. 2009). MiR-128a directly targets the Bmi-1 oncogene (polycomb ring finger oncogene; BMI1), which increases p16INK4A expression and reactive oxygen species (ROS). Collectively, these effects promote cellular senescence in medulloblastoma cell lines. MiR-217, which is expressed in endothelial cells during aging, promotes premature senescence by inhibiting SIRT1 expression. This occurrence increases forkhead box O1 (FoxO1) expression (Garzon et al. 2008). In addition, miR-217 has been reported to be a novel tumor suppressive miRNA that targets K-Ras in pancreatic ductal adenocarcinoma due to decreases in tumor cell growth both in vitro and in vivo (Menghini et al. 2009). MiR-20a induces senescence in MEFs by directly downregulating the transcriptional regulator leukemia/lymphoma-related factor (LRF), which induces p19ARF (Borgdorff et al. 2010). In addition, miR-519 induces senescence in cancer cell lines by repressing HuR expression (Voorhoeve et al. 2006). In contrast, there are miRNAs that are downregulated during senescence, such as miR-15b, miR-24, miR-25, and miR-141, which directly target mitogen-activated protein kinase kinase (MKK4) (Cho et al. 2009). Recently, it was shown that 28 miRNAs prevented senescence induced by oncogenic RasG12V (Borgdorff et al. 2010). These miRNAs bypass RasG12V-induced senescence by directly targeting the 3′UTR of p21Cip1. Moreover, miR-372, miR-373, miR-302, and miR-520 also bypass RasG12V-induced senescence through the downregulation of LATS2 in addition to p21Cip1 (Borgdorff et al. 2010). These identified proliferative miRNAs are associated with cancer development (Leal et al. 2013; Feliciano et al. 2011).
Over all steps, DNA methylation regulates expression of senescence genes, with the capability of controlling the process (Carnero and Lleonart 2010). In human cancers, the silencing of tumour suppressor genes through aberrant DNA methylation of the CpG island(s) in promoters in these genes is a common epigenetic change (Baylin et al. 2000). There are an assortment of pathways from which genes have been shown to be hypermethylated in cancer cells, including DNA repair, cell-cycle control, invasion and metastasis. The tumour suppressor genes BRCA1, p16INK4a, p15INK4b, p14ARF, p73 and APC are among those silenced by hypermethylation, although the frequency of aberrant methylation is somewhat tumour-type specific. Recently, we found S-adenosylhomocysteine hydrolase (SAHH) (Leal et al. 2008a), which has also been previously identified in an independent short hairpin RNA (shRNA) screening (Brummelkamp et al. 2004), the inactivation of which confers resistance to both p53- and p16(INK4)-induced proliferation arrest and senescence. SAHH catalyzes the hydrolysis of S-adenosylhomocysteine to adenosine and homocysteine. In eukaryotes, this is the major route for disposal of S-adenosylhomocysteine formed as a common product of each of the many S-adenosylmethionine-dependent methyltransferases, therefore regulating the methylation processes. Interestingly, SAHH inactivation inhibits p53 transcriptional activity and impairs DNA-damage-induced transcription of p21(Cip1). SAHH messenger RNA (mRNA) was lost in 50 % of tumour tissues from 206 patients with different kinds of tumours in comparison with normal tissue counterparts. Moreover, SAHH protein was also affected in some colon cancers (Leal et al. 2008a; Castro et al. 2006).
7.3 Clinical Implications
The implication of senescence as a barrier to tumorigenesis first comes from the realisation that a limited number of duplications necessarily reduces the possibility of tumor growth. However, the proliferative lifespan before reaching the Hayflick limit could be sufficient to generate a tumor mass greater than that required for lethality. This argument fails to take into account the existence of ongoing cell death and differentiation within a tumor and the occurrence of clonal selection driven by different senescence barriers or barriers unrelated to senescence. Finally, a clinically significant cancer can be composed of entirely mortal, pre-senescent cells if the cell of origin has a sufficient proliferative lifespan and the tumor develops with few successive clonal expansion steps and/or with a low cell death rate. Even with these examples, however, senescence may of course still be a significant barrier to the recurrence of tumors from the small number of residual cells remaining after therapy.
As mentioned, several studies in vivo show that oncogene-induced senescence provides a bona-fide barrier to tumorigenesis. Michaloglou and co-workers (Michaloglou et al. 2005) have shown that an oncogenic BRaf can induce senescence in fibroblasts and melanocytes and that human nevi display markers of senescence. Therefore, sustained exposure of melanocytes to aberrant mitotic stimuli provokes senescence after an initial proliferation burst. Collado and co-workers (Collado et al. 2005) identified senescent cells in vivo after generating new senescence biomarkers from array studies. Using conditional Kras-val12 mice strains they observed senescence markers to be predominant in premalignant lesions of the lung and pancreas, but not in those that have progressed to full-blown cancers. Direct evidence that hyperproliferative signals can trigger a program of permanent arrest in vivo have been provided in a transgenic model conditionally expressing E2F3 in the pituitary gland (Lazzerini Denchi et al. 2005). E2F3 induced hyperplasias that failed to progress because the cells became insensitive to further mitogenic signals. This insensitivity correlated with the appearance of senescence markers and a terminally arrested cellular state. Disruption of PTEN in mice also produces hyperplastic conditions analogous to prostatic intraepithelial neoplasia (a precancerous lesion in men). These lesions display senescence markers (Chen et al. 2005). Loss of p53 prevents senescence in response to PTEN ablation and cooperates to produce invasive prostate carcinomas. These results are consistent with the notion that senescence actively limits malignant conversion.
In human fibroblasts in culture, the senescence program involves chromatin reorganisation involving H3 methylation at the Lys9 residue concomitant with the recruitment of heterochromatin proteins to some proliferation-related genes. Braig and co-workers (Braig et al. 2005) found that disruption of Suv39h1 methyltransferase, which methylates the Lys9 residue of H3, blocked ras-induced senescence and accelerated ras-induced lymphomagenesis in mice. Interestingly, Suv39h1-expressing tumors responded through senescence to chemotherapy; however, Suv39h1-null tumors did not show any senescent response but still maintained the apoptotic response. Treating ras transgenic mice with DNA-methyltransferase or histone deacetylase inhibitors, which mimic the effects of Suv39h1 disruption, accelerated ras-induced tumorigenesis.
The concept of cancer being a disease whereby cells have lost the ability to senesce leads to a critical evaluation of the benefits that can be achieved for cancer diagnosis and therapy through the knowledge surrounding molecular pathways (both genetic and epigenetic in origin) that induce senescence. Until just a few years ago, it was accepted that tumor cells were no longer capable of senescence. Today, however, it is accepted that neoplastic cells can be forced to undergo senescence by genetic manipulations and by epigenetic factors, including anticancer drugs, radiation and differentiating agents (Carnero et al. 2003; Roninson 2003). However, although not fully studied in vivo, it has been shown that senescent cells might increase the oncogenic potential of tumor cells. Therefore it will be necessary to understand the contribution of senescent stromal cells to tumors, before applying drug-induced senescence program to tumors.
Immortalising defects are recessive and can be blocked by imposing the process of senescence (Pereira-Smith and Smith 1988). The first approach to inducing senescence to tumor cells was through somatic cell fusion. These studies identified four senescence-determining complementation groups. In recent years, it has been found that different tumoral cell lines show cellular growth arrest along with senescence markers after the genetic expression of tumor suppressor genes commonly involved in senescence, such as p53, p21, p16, pRb or p21 (McConnell et al. 1998). Similarly, the restoration of cellular levels of p53 in a cell line conditionally immortalised by p53 antisense expression induces growth arrest with a senescent phenotype (Carnero et al. 2000a). Adenovirus vectors carrying CKIs (p16INK4a, p15INK4b, p21cip1 and p27kip1) as vehicles for delivery and expression are a powerful approach to examining therapeutic applications both in vitro and in vivo, with promising results (Carnero 2002). When a 16-amino acid transmembrane carrier segment derived from the Drosophila antenappedia protein was linked to the third ankyrin repeat of the p16INK4a protein and inserted into cells, Rb-dependent G1 arrest was observed. In a breast-derived cell line, the chimera containing the antennapedia peptide and the carboxyl-terminal residue of p21waf1 had higher specificity for CDK4/cyclin D than for CDK2/cyclin E and arrested the cells in G1 phase (Ball et al. 1997).
These observations indicate that tumor cells maintain at least some of the components of the cellular senescence program, including terminal growth arrest. It is now clear that, depending upon the cell proliferation kinetics of the tissue of origin, tumor development can be initiated by genetic events, causing either a block in terminal differentiation or/and inappropriate activation of growth stimulatory signaling pathways. The net result in both cases is the generation of a cellular clone capable of infinite expansion if it is not constrained by physical barriers or lack of blood supply. Lowe and collaborators (Schmitt et al. 2002) convincingly showed that in a lymphoid mouse tumor model, an intact senescence pathway appears to be pivotal to the efficacy of cyclophosphamide, and its disruption makes tumor cells highly refractory to the drug. On the other hand, as mentioned, Suv39h1-expressing tumors responded to chemotherapy by inducing senescence. However, Suv39h1- null tumors did not show any senescent response but still maintained the apoptotic response. Suv39h1-null tumors with altered apoptotic response do not respond to therapy.
These results suggest that drug efficacy and tumor formation are not fully independent processes. Until recently, tumor formation and the development of drug resistance were thought to be independent processes. Mutations in factors that regulate tumor-suppressive fail-safe mechanisms, such as apoptosis and senescence, allow transformation. Chemotherapeutic compounds activate a separate set of effector pathways that eliminate malignant clones. Mutations in factors that are involved in these separate pathways inhibit the effect of chemotherapy to induce the effector programs to eliminate the tumors. Consequently, defects in antineoplastic fail-safe programs, even if required to allow for tumor formation, do not interfere with the effector program initiated by therapeutic agents. Nevertheless, preclinical data have provided evidence that key regulators, such as p53, participate in tumor prevention and drug action, and that tumor mutations acquired during tumor development also confer chemoresistance (Lowe et al. 1993, 2004).
The in vitro observation that DNA-damaging agents not only promote apoptosis but also induce cellular senescence (Roninson 2002; te Poele et al. 2002) indicates that genes that control senescence might also determine treatment outcome. Using a MYC-driven mouse lymphoma model, p53 and p16INK4A were recently shown to control drug-induced senescence in vivo (Schmitt et al. 2002). Drug-treated lymphomas with apoptotic defects were forced into senescence, and tumors that resumed growth frequently displayed defects in either p53 or p16INK4A. Importantly, drug-induced senescence was shown to contribute to long-term host survival after cancer therapy, as mice bearing lymphomas that were unable to enter senescence in response to therapy had shorter survival times. Notably, drug-inducible senescence is not a phenomenon that is restricted to a mouse lymphoma model, as tissue specimens taken from human breast tumors after chemotherapy also displayed typical features of cellular senescence (te Poele et al. 2002).
Depending on the initiating oncogene, transformation relies on fail-safe defects that disrupt either apoptosis or senescence. There are a number of reports that drug-inducible senescence could become detectable only after apoptosis has been disabled (Maloney et al. 1999). It is conceivable that senescence occurs with much slower kinetics, serving as a ‘backup’ fail-safe program in case the first-line response is corrupted. This is supported by sequential disruption of apoptosis- and senescence-controlling genes during tumor formation and subsequent therapy reported in human cancers (Carter et al. 2001; Elenitoba-Johnson et al. 1998).
7.4 Senescence Based Therapy
Different chemical agents can induce cellular senescence epigenetically. Treatment of primary cells with H2O2 or butyrate provokes early senescence (Chen 2000). Similar results were obtained after treatment with high doses of radiation and other damaging agents (Chen 2000). Interestingly, the treatment of different tumor cell lines with different chemotherapeutic agents, radiation or differentiating agents induces irreversible growth arrest, with enzymatic and morphologic changes resembling those occurring during replicative senescence. Moderate doses of doxorubicine induced a senescent phenotype in 11 out of 14 tumor cell lines analysed, independently of p53 status (Chang et al. 1999a). A similar effect has been observed in lines from human tumors treated with cisplatin (Wang et al. 1998b), hydroxyurea (Yeo et al. 2000) and bromodeoxyuridine (Michishita et al. 1999). In mammary carcinoma cell lines treated in vitro and in vivo with differentiating agents, terminal proliferative arrest with minimal toxicity for normal cells has been observed (Chang et al. 1999b).
The propensity of tumor cells to undergo senescence in response to different kinds of damage induced by commonly used chemotherapeutic treatments was compared on cell lines from different tumor origins Elenitoba-Johnson et al. 1998. Under equitoxic doses, the strongest induction of a senescent phenotype was observed with DNA-interacting agents (doxorubicin, aphidicolin and cisplatin) and the weakest effect was observed with microtubule-targeting drugs (Taxol and vincristine). A medium response was observed with ionising radiation, cytarabine and etoposide. Induction of senescence by the drugs was dose dependent and correlated with the growth arrest observed in the cultures (Chang et al. 1999b; Chen 2000; Michishita et al. 1999; Yeo et al. 2000). The drug-induced senescent phenotype in tumor cells was not associated with telomere shortening and was not prevented by the expression of telomerase (Elmore et al. 2002).
Drug-induced senescent phenotypes have been confirmed in vivo (Schmitt et al. 2002). A study from Poele et al. (2002) revealed the correlation between chemotherapeutic treatment in clinical cancer and the senescence response. In frozen samples from breast tumors treated by neoadjuvant chemotherapy (cyclophosphamide, doxorubicin and 5-fluoracyl), senescent markers were detected in 41 % of samples from treated tumors. Normal tissue was negative, suggesting that the chemotherapy-induced senescence was a specific response of tumor cells. Interestingly, senescence response was associated with wild type p53 and the increased expression of p16. Similarly, in treatment-induced senescence, murine Eμ-myc lymphoma response required wild type p53 and p16 (Schmitt et al. 2002). It is interesting to explore whether the efficacy of current therapeutic regimes applied to tumors depend on the de novo senescence-induced phenotype, and whether there is any difference according to the tissue of origin, i.e. Mesenchymal vs epithelial.
The Chk2 kinase is a tumor suppressor and key component of the DNA damage checkpoint response that encompasses cell cycle arrest, apoptosis, and DNA repair. It has also been shown to have a role in replicative senescence resulting from dysfunctional telomeres. Some of these functions are at least partially exerted through activation of the p53 transcription factor. High-level expression of Chk2 in cells with wild type p53 led to arrested proliferation with senescent features (Gire et al. 2004). These were accompanied by p21 induction, consistent with p53 activation. However, Chk2-dependent senescence and p21 transcriptional induction also occurred in p53-defective cells. Small interfering RNA-mediated knockdown of p21 in p53-defective cells expressing Chk2 resulted in a decrease in senescent cells. DNA-damage response is also induced by cytokines, such as interferons. Sustained treatment with interferon triggers a p53-dependent senescence program. Interferon-treated cells accumulated gamma-H2AX foci and phosphorylated forms of ATM and CHK2. The DNA damage signalling pathway was activated by an increase in reactive oxygen species (ROS) induced by interferon and was inhibited by the antioxidant N-acetyl cysteine. RNA interference against ATM inhibited p53 activity and senescence in response to beta-interferon (Moiseeva et al. 2006). It seems that p53 activation is the primary response to DNA damage, but its absence does not preclude a response with a senescent phenotype.
Comparable to p53, which functions as a fail-safe mediator of DNA-damage response, the p16 inhibitor has been implicated in both response to DNA-damage and control of stress-induced senescence. Although the molecular mechanism used by p16 to control not only temporary but permanent cell cycle arrest is unclear, p16 responds to DNA-damage in a delayed manner and appears to be indispensable for the maintenance of cellular senescence (Schmitt et al. 2002; te Poele et al. 2002). A synthetic inhibitor of CDK4/CDK6, possibly mimicking the role of p16, produced a DNA-damage-independent form of senescence in cells lacking p16 expression and inhibited the growth of tumors in mice (Roberts et al. 2012). This inhibitors is being shown to produce great effect in human clinical trials (Flaherty et al. 2012), but at present we do not know if this efficacy is caused by the CDK4/cell cycle inhibition or by the senescence induction.
Use of siRNAs to inactivate the papilomavirus oncoproteins E6 and E7, which deregulate p53 and pRb, restored cellular senescence in cervical cancer cells. Introduction of E2 protein, a negative regulator of E6 and E7, induced senescence in almost all cervical carcinoma cells tested. The effect of E2 was not accompanied by telomere shortening, nor was it prevented by telomerase expression. Induction of senescence by E2 was associated with p53 stabilisation and strong induction of p21, and it was prevented by using p21 antisenses (Wells et al. 2000).
Many observations indicate that p53, p21 and p16, which regulate cellular senescence, play an important role in treatment-induced senescence of tumor cells. Since these genes are commonly lost in human tumors, we can expect that most human tumors do not respond by undergoing senescence. However, this is not the case. Chemotherapeutic drugs induced senescence in p53- and p16-defective tumor cell lines (Chang et al. 1999b). In vivo, 20 % of tumors undergoing senescence after treatment showed p53 mutations (te Poele et al. 2002). We have been able to induce senescence with several chemotherapeutic drugs in p53-null cells independently of p16 (Moneo and Carnero, unpublished). We have found that the induced senescence correlated with p53-independent p21 induction. Moreover, knock-out of p53 or p21 in HCT116 cells decreased but did not abolish cellular senescence. Hence, p16, p53 and p21 might acts as positive regulators but are not absolutely required for this response. Other related tumor suppressors, such as p63 or p73, could be involved, and their role in drug-induced senescence should be explored.
Related posts:
 Introduction to Cell Stress Responses in Cancer: The Big Picture
Introduction to Cell Stress Responses in Cancer: The Big Picture
 Targeting Hypoxic Adaptations of Cancer Cells: Molecular Mechanisms and Therapeutic Opportunities
Targeting Hypoxic Adaptations of Cancer Cells: Molecular Mechanisms and Therapeutic Opportunities
 Inflammatory Dysregulation and Cancer: From Molecular Mechanisms to Therapeutic Opportunities
Inflammatory Dysregulation and Cancer: From Molecular Mechanisms to Therapeutic Opportunities
 Cell Cycle Checkpoint and DNA Damage Response Defects as Anticancer Targets: From Molecular Mechanisms to Therapeutic Opportunities
Cell Cycle Checkpoint and DNA Damage Response Defects as Anticancer Targets: From Molecular Mechanisms to Therapeutic Opportunities
 The Hsp70 Family of Heat Shock Proteins in Tumorigenesis: From Molecular Mechanisms to Therapeutic Opportunities
The Hsp70 Family of Heat Shock Proteins in Tumorigenesis: From Molecular Mechanisms to Therapeutic Opportunities
 Cell-Nonautonomous ER Stress-Mediated Dysregulation of Immunity by Cancer Cells
Cell-Nonautonomous ER Stress-Mediated Dysregulation of Immunity by Cancer Cells
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


