Keywords
HSV, oncolysis, immune response, therapeutic transgene, safety, vector delivery, glioma
Introduction
Herpes simplex virus 1 (HSV-1) is a large, naturally neurotropic, double-stranded DNA virus. Primary infection with wild-type HSV-1 occurs at the oral and genital mucosa. Virus is then retrogradely transported from sensory nerve endings to regional sensory ganglia, where the virus replicates or becomes latent through the formation of a stable episomal element . Viral reactivation during episodes of relative immunosuppression leads to a spectrum of clinical conditions ranging from relatively mild and common oral and genital ulceration to life-threatening encephalitis. It has been demonstrated that up to 80% of some populations are seropositive to HSV-1 .
HSV-1 has a large genome of 150 kb, encoding approximately 80 proteins. Based on the microenvironment of the infected cell, viral infection proceeds along one of two pathways. Either active gene expression occurs and new viral particles are formed, leading to cell lysis and the infection of adjacent cells, or the majority of viral gene expression is suppressed and long-term, latent infection occurs . Generally speaking, these two pathways of infection have been exploited to develop HSV-1-based vectors with distinctly different properties:
- 1.
Recombinant replication-defective HSV-1 vectors that exploit HSV latency and neuron-specific promoters to achieve long-term transgene expression in the brain.
- 2.
Recombinant replication-selective HSV-1 vectors that are capable of lytic infection (ideally in tumor cells) in the presence of appropriate gene products supplied in trans . These vectors have been developed for the treatment of a range of cancers and are the subject of this chapter.
HSV-Derived Oncolytic Viruses
HSV has a number of properties that make it ideal for its use as an oncolytic vector, including a broad tropism, a potent cytolytic potential, and a stable, nonintegrating genome containing numerous nonessential genes. This allows for the insertion of multiple therapeutic transgenes encoded in up to 30 kbp. In addition, there are antiviral agents that can be administered to patients to effectively prevent further viral replication in the event of excessive treatment toxicity.
Mutations in a number of different genes have been shown to selectively limit HSV-1 replication to dividing cells. These mutations have been exploited in the development of replication-selective HSV-1 vectors for the treatment of tumors. To date, two replication-selective viral constructs have reached clinical trials in patients with primary brain tumors. These viruses, designated G207 and HSV1716, harbor null mutations in both copies of the γ 1 34.5 gene. The product of this gene, ICP34.5, has a number of important functions, which are critical in enhancing the ability of HSV-1 to infect neurons and overcome host cell responses to viral infection.
Normally, in noncancerous cells, viral infection with HSV-1 leads to protein kinase R (PKR) upregulation. This leads to phosphorylation and subsequent deactivation of eukaryotic initiation factor-2α (EIF2α), causing shutdown of translation and therefore limited viral replication. ICP34.5 increases HSV-1 virulence by binding to protein phosphatase 1α (PP1α), causing dephosphorylation of eIF2α, increasing protein synthesis, and thus leading to a favorable environment for viral replication . As a consequence, HSV-1 vectors that harbor null mutations in both copies of the γ 1 34.5 gene are capable of replicating only in cells with disrupted PKR pathways . Indeed, there is evidence that the ability of these viral constructs to lyse tumor cells relates to the overexpression of the ras oncogene, which is known to disrupt the PKR pathway . Furthermore, deletions of the γ 1 34.5 gene remove sequences of the latency-activated transcripts that are encoded in the complementary antisense strands of DNA, limiting the ability of these viruses to establish latency .
In addition to null mutations in both copies of the γ 1 34.5 gene, the G207 HSV-1 vector has a deletion mutation in the U L 39 gene. This gene encodes the large subunit of viral ribonucleotide reductase (ICP6), which is required for nucleotide synthesis in terminally differentiated cells such as neurons. Because dividing cells effectively supply this enzyme in trans , the ability of G207 to replicate only in tumor cells is enhanced . Usefully, this deletion also makes G207 more sensitive to the antiviral effects of ganciclovir, enhancing its safety profile . Additional deletions of the α47 gene, the product of which inhibits antigen presentation, and the overlapping US11 promoter region, which places the US11 gene under the control of the immediate-early α47 promoter, thus promoting viral replication, have been undertaken to create an oncolytic vector termed G47Δ . This vector has been shown to be more efficacious than G207 in a variety of animal tumor models .
A key limitation of vector constructs harboring mutations in the γ 1 34.5 gene is that the resulting impairment of protein synthesis and replication may reduce their oncolytic potential. A number of strategies to overcome this limitation have been developed and shown to enhance tumor cell kill in preclinical studies. These strategies have included the use of tumor-specific promoter sequences such as nestin or B-myb to facilitate intratumoral γ 1 34.5 gene expression. Further attempts to enhance treatment efficacy and minimize damage to normal cells have included the development of HSV targeted to specific receptors overexpressed by a large proportion of tumor cells. Specifically, vectors have been developed that are targeted to a mutant form of the human epidermal growth factor receptor 2 (EGFRvIII) and the IL13α2 receptor . Alternative strategies have included the development of chimeric HSV vectors expressing the PKR-evasion genes TRS1 or IRS1 from the human cytomegalovirus, which restores late viral protein synthesis and subsequently oncolysis without increasing neurotoxicity . Of these latter chimeric HSV/HCMV vectors, C134, which harbors deletions in both copies of the γ 1 34.5 gene and contains the IRS1 gene, is scheduled for evaluation in a forthcoming clinical trial.
Immune Responses to Oncolytic HSV
HSV is highly efficient at evading the immune system through a number of diverse mechanisms, including the following:
- 1.
The envelope glycoprotein heterodimer gE-gI binds to and masks the F C domain of IgG, preventing complement activation and phagocytosis .
- 2.
The envelope glycoprotein C (gC) binds complement factor C3, preventing complement activation .
- 3.
The tegument protein virion host shutoff (vhs) disrupts host cell protein synthesis, significantly reducing major histocompatibility complex-1 (MHC-1) expression, limiting the ability of cytotoxic T cells to recognize and destroy HSV-infected cells .
- 4.
The immediate-early gene product of ICP47 impedes viral antigen presentation via MHC-1 .
It is important to note that despite the ability of wild-type HSV to evade the immune system, up to 90% of the human population has antibodies to HSV, most commonly toward the envelope glycoproteins gB, gC, and gD . Importantly, in a syngeneic intracranial rat glioma model, the transduction efficiency of an HSV-1 vector construct harboring a deletion mutation of ribonucleotide reductase showed an 85% decrease in transduction efficiency in preimmunized animals compared to controls , although in another study, preexisting immunity did not affect the extent of the observed antitumor response in a murine lymphoma model . Nevertheless, this raises concerns regarding the use of HSV-1-based vectors in clinical practice, particularly due to the high incidence of immunity in the human population. Furthermore, although wild-type HSV is clearly adept at evading the immune system to cause latent infection in naive subjects, this effect is likely to be significantly impaired by the deletion of key genes, such as ICP47 and vhs, from HSV-derived oncolytic vectors.
HSV-mediated oncolysis precipitates a profound inflammatory response with potential activation of the adaptive immune system against the tumor. Indeed, this antitumor immune response may be critical in the therapeutic efficacy observed with oncolytic HSV constructs in preclinical and some clinical studies, and this effect significantly enhances the practicality of using oncolytic vectors for the treatment of tumors with widespread metastases. This is highlighted by the regression of tumors distant from a tumor injected with oncolytic HSV in a rodent colon carcinoma model , as well as inflammation of uninjected cutaneous and subcutaneous tumor deposits in a clinical trial following injection of an oncolytic viral vector expressing granulocyte–macrophage colony-stimulating factor (GM-CSF) . The mechanism underlying this response is unclear, although it is likely to require dendritic cell uptake of tumor antigen and presentation to T cells. Indeed, there is evidence that oncolytic HSV infection promotes uptake of antigen by dendritic cells and possible dendritic cell maturation . This may be a critical step leading to the migration of natural killer cells and CD8 + T cells into the tumor following intratumoral injection of oncolytic HSV .
Whereas the relationships between treatment with oncolytic HSV and the occurrence of an adaptive antitumor immune response are being elucidated, the relevance of this adaptive immune response in the context of treating primary malignant brain tumors is less clear, particularly because the brain exhibits a state of relative immune privilege. Nevertheless, because oncolytic HSV infusion into the brain induces an intense CD8 + T cell infiltration, it seems feasible that oncolyic HSV may provoke an adaptive antitumor immune response in the brain as well. Clearly, more research is required to characterize the nature and significance of immune responses to intracranially administered HSV-based vectors before these vectors can be reliably used in clinical trials. Of particular urgency, in view of the high incidence of antibodies to HSV in the human population, is the need to establish the effects of preexisting immunity on HSV vector-mediated transgene expression and oncolysis.
Therapeutic Transgenes
In addition to the ability to selectively replicate in and lyse tumor cells, HSV vectors have been developed that take advantage of their large genome to deliver a range of therapeutic transgenes. Theoretically, this “arming” of oncolytic HSV vectors has the potential to minimize the vector dose required to demonstrate treatment efficacy, although theoretically effective oncolysis with release of therapeutic transgene products into the extracellular space is likely to increase treatment toxicity and may even impair viral replication. Previously, development of armed oncolytic HSV vectors was an extremely time-consuming process. In recent years, development of T-BAC and HSVQuik systems based on G47Δ- and G207-like backbones , respectively, has significantly improved the practicality of generating armed oncolytic vectors. In general, four types of transgenes have been employed in preclinical studies to arm oncolytic HSV.
Suicide Genes
The use of suicide genes to arm oncolytic viruses offers a number of significant advantages. First, they enhance vector-mediated toxicity to the transduced cell. In addition, following cell lysis, suicide gene products may diffuse through the extracellular space, exerting toxic effects on bystander cells. However, although these cells may be tumor cells, in the context of highly infiltrative tumors such as malignant gliomas, they may also be normal cells, exacerbating potential treatment toxicity. Furthermore, suicide gene products have the clear potential to limit cell replication, thereby potentially reducing treatment efficacy. Consequently, there is a clear requirement for optimized temporal regulation of oncolysis and therapeutic transgene expression.
HSV-1 expresses the prodrug-activating gene thymidine kinase naturally. This enzyme phosphorylates intravenously administered ganciclovir (GCV) to GCV monophosphate. This is further phosphorylated by endogenous cellular enzymes to GCV triphosphate and incorporated into replicating DNA by DNA polymerase. The incorporation of GCV triphosphate prevents DNA chain elongation, promoting cell death. However, despite the frequent use of this enzyme/prodrug combination in preclinical studies and clinical trials, there are significant limitations to its use in the treatment of brain tumor in particular. First, the antitumor effects of GCV triphosphate are restricted to replicating cells, which for high-grade gliomas represent 14–44% of the tumor cell population . Second, systemically administered ganciclovir has very limited penetration across the blood–brain barrier due to its low lipophilicity. Indeed, animal studies examining the biodistribution of ganciclovir, following intravenous administration, have demonstrated peak brain tissue concentrations of just 2.5% of the peak plasma concentration and a brain tissue half-life of only 1 hr . Furthermore, the maximum achievable plasma concentration of ganciclovir in clinical practice is limited by toxicity—most commonly neutropenia, which affects approximately 40% of patients .
In view of the limitations of thymidine kinase as a prodrug activating gene, a number of alternative genes that promote cell death have been used to arm oncolytic viral vectors. These genes include rat cytochrome P450 2 B1, human intestinal carboxylesterase, and cytosine deaminase, which are capable of converting cyclophosphamide , irinotecan , and 5-fluorocytosine into active metabolites. Although these suicide genes have yet to be employed in clinical trials, there is evidence from these preclinical studies that they enhance antitumor effects without impairing viral replication.
Immunomodulatory Genes
As previously discussed, there is a close interplay between immune responses directed against oncolytic HSV and any antitumor effects observed . As a consequence, the use of therapeutic transgenes that augment the antitumor immune response without impairing viral replication is a rational approach to enhancing antitumor efficacy. Immunomodulatory therapeutic transgenes that have been employed in preclinical studies to arm oncolytic HSV vectors and that have been demonstrated to have an enhanced antitumor response compared to parental virus in a variety of tumor models include interleukin (IL)-4, IL-12 , IL-18 , CD40 ligand , 6CK , CCL2 , and soluble B7-1 . This effect is likely related to the significantly increased influx of lymphocytes and macrophages into the tumor when these transgenes are employed. In addition, a vector armed with tumor necrosis factor-α (TNF-α), which is known to lead to tumor cell apoptosis, has been developed under the influence of a late promoter sequence, minimizing the effects of TNF-α on viral replication .
To date, the only armed oncolytic HSV that has been employed in a clinical trial harbors a deletion of the α47 gene and has the γ34.5 gene replaced with GM-CSF. This vector (OncoVEX GM-CSF ) has been injected into subcutaneous deposits of breast, gastrointestinal, and head and neck malignancies and into recurrent melanomas, and it has been demonstrated to lead to intense local inflammatory responses with evidence of tumor necrosis. Furthermore, in keeping with induction of antitumor immunity, inflammation was observed in distant, uninjected tumors .
Anti-Angiogenic Genes
Wild-type herpes simplex virus infection leads to enhanced tissue vascularity. This effect is mediated by the suppression of smooth muscle and endothelial cell release of anti-angiogenic proteins such as thrombospondin-1 and -2 and possible upregulated expression of vascular endothelial growth factor and matrix metalloproteinase-9 (MMP-9) . This effect is preserved following in vivo injections of HSV G207 but not G47Δ, and it is clearly of significant concern because iatrogenic enhancement of tumor vascularity has the potential to lead to tumor growth. However, G47Δ vectors armed with anti-angiogenic genes such as platelet factor-4 or dominant-negative fibroblast growth factor receptor effectively inhibit tumor angiogenesis and growth in animal models of glioma and nerve sheath tumors . In addition, arming oncolytic HSV with an inhibitor of MMP-3 inhibits the growth of neuroblastoma and peripheral nerve sheath tumors in animal models and is associated with reduced vascular density .
Fusogenic Proteins
In order to enhance the tumor cell kill, oncolytic vectors have been armed to express fusogenic proteins such as the gibbon ape leukemia virus fusogenic glycoprotein ( GALV.fus ). Fusion of infected cells with surrounding cells leads to the formation of a syncytium, in which viral replication and potentially coexpression of other therapeutic transgenes may occur. The use of GALV.fus has been shown to enhance the antitumor effects of oncolytic HSV vectors in models of hepatocellular carcinoma and malignant glioma .
Vector Delivery
A frequently overlooked aspect in the application of gene therapy in clinical practice is the use of appropriate delivery strategies. In preclinical and clinical trials of oncolytic HSV, vector delivery has typically been achieved using direct intratumoral injection, although other delivery strategies, including intravascular vector injection, have been employed in a number of preclinical studies and clinical trials. However, in view of the high rate of preexisting immunity to HSV in the human population and the likelihood that only a very small proportion of tumor cells will be infected following intravascular delivery, combined with the likelihood of extensive vector uptake in organs such as the liver and spleen, it seems unlikely that this approach will prove to be efficacious or safe in clinical practice. Realistically, highly targeted vector delivery is likely to be an essential prerequisite to the use of oncolytic HSV in clinical trials. In this regard, the administration of HSV in clinical trials of patients with malignant gliomas is instructive.
Malignant gliomas are highly infiltrative tumors. Whereas the main tumor mass can often be treated effectively with surgery and/or radiotherapy, targeted eradication of the infiltrating tumor cells is technically challenging. The efficacy of vector-mediated oncolysis depends in part on transducing as many tumor cells as possible on initial vector administration. It is likely that tumor tissue architecture, particularly in the form of tissue necrosis and neovascularity, may impede the ability of HSV to replicate and spread through a tumor. Indeed, there is evidence that oncolytic HSV dissemination is diminished in collagenous areas of tumor, and tissue pre-infusion with agents capable of degrading extracellular matrix, such as collagenase and MMPs , has been demonstrated to enhance oncolytic HSV distribution and efficacy. However, because malignant gliomas contain large volumes of tissue necrosis, direct intratumoral vector injection is unlikely to lead to oncolysis throughout the entire tumor volume. This is reflected in the failure of previous clinical trials to demonstrate convincing evidence of efficacy in the treatment of high-grade gliomas . Consequently, the focus of recent studies has been on the injection of replication-selective HSV vectors into peritumoral brain .
Injection Versus Convection-Enhanced Delivery
In preclinical studies, viral vector delivery to the brain has been achieved by direct injection into the parenchyma, the ventricular system, and the thecal sac. Despite the use of direct intraparenchymal injection in a number of clinical trials and the use of all of these techniques extensively in preclinical studies, there is very little evidence for their effectiveness in achieving widespread vector distribution. For example, cell transduction following intraventricular adenoviral vector injection has been shown to be limited to ependymal cells and following intraparenchymal injection to be limited to an area no more than a few hundred micrometers from the needle-tip . Intrathecal viral vector-mediated lysosomal enzyme delivery has been attempted in mouse models of type I and VII mucopolysaccharidoses with both lentivirus and recombinant adeno-associated viral (AAV) vectors . Despite the small size of the mouse brain, cell transduction was again limited to areas of the brain in close proximity to the CSF, such as the olfactory bulbs and cerebellum. Furthermore, Passini et al . were able to achieve very limited AAV-mediated neuronal transduction with β-glucuronidase following intraventricular viral injection with rAAV (serotypes 2 and 5) in β-glucoronidase-deficient neonatal mice .
In view of the limited distribution seen in the majority of studies employing direct injection and the massive difference in brain volume between humans and animals frequently used in research, such as rodents and primates, it is highly unlikely that these injection-based approaches can achieve adequate vector distribution in clinical practice. Indeed, it has been estimated that to administer AAV by direct intraparenchymal injections to an entire infant brain would require 368 injections , which clearly would be neither safe nor practical.
Convection-enhanced delivery (CED) involves the implantation of fine catheters, with an outer diameter typically less than 1 mm, directly into the brain or spinal cord. Infusion of drugs through these catheters at precisely controlled, low-infusion rates (0.1–10 μl/min) leads to bulk flow of drug into the brain extracellular space and distribution over potentially large volumes of tissue ( Figure 14.1 ) . Through accurate positioning of the catheter tip and the use of therapeutic agents such as oncolytic HSV that are unable to cross the blood–brain barrier, it may be possible to compartmentalize drugs within discrete neuroanatomical structures, limiting the risk of systemic toxicity. This clearly offers tremendous advantages in the delivery of oncolytic HSV to the very large volumes of brain that would be required to treat highly infiltrative malignant brain tumors.
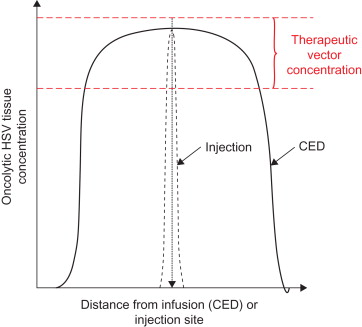
Convection-Enhanced Delivery of HSV
Because it is possible to achieve widespread vector distribution in the brain by CED, this represents the most rational approach for the delivery of oncolytic HSV vectors to these areas of tumor-infiltrated brain in clinical practice. However, despite the large number of preclinical studies that have involved the direct intracranial administration of HSV-based vectors, it is questionable whether HSV-based vectors, with a diameter of between 120 and 300 nm, can be infused through the brain extracellular space, which has a diameter of up to 80 nm. Indeed, following CED-based infusions of HSV into the gray and white matter of normal rats, we demonstrated that vector infusions at 0.5 µl/min led to extensive tissue damage in both gray and white matter with minimal penetration of vector into the tissue. Infusions at higher flow rates and therefore with a greater pressure gradient caused similar levels of tissue damage and poor vector distribution . This tissue damage draws parallels with postmortem observations demonstrating cystic cavities at the infusion site in the brain of two patients who had HSV1716 injected intratumorally .
Because heparan sulfate proteoglycans are known to act as cell surface receptors for HSV-1 as well as a number of other viruses, including serotype 2 recombinant adeno-associated virus (rAAV2), we hypothesized that co-infusion with heparin may facilitate HSV distribution. Indeed, co-infusion of heparin at a concentration of up to 5000 IU/ml has been shown to improve the distribution of rAAV2 in the striatum of adult rats, presumably by competitive antagonism of viral binding to heparan sulfate proteoglycans . However, in our study, co-infusion of HSV at a concentration of 5000 IU/ml led to a slight increase in HSV distribution but with associated extensive intracerebral hemorrhages in both gray and white matter . As such, this approach is unlikely to be of significant value in clinical practice.
A number of studies have evaluated the CED-based distribution properties of nanoparticles of a similar size to HSV-1. For example, polystyrene nanoparticles and liposomes with a diameter of 200 nm only penetrate short distances into the striatum of rats . Coating these nanoparticles with albumin to shield the hydrophobic particle surface significantly improved their distribution . We combined these approaches by pre-infusing both white matter and gray matter with isotonic albumin solutions to shield nonspecific binding and achieve isotonic expansion of the brain extracellular space to facilitate the administration of HSV. This approach facilitated the widespread distribution of vector in the white matter of rat and pig brain, although it was unsuccessful in the gray matter of rats . The ability to distribute HSV following tissue pre-infusion in white matter and not gray matter probably relates to the greater elasticity of white matter and subsequently its greater capacity to accommodate an infused volume . Indeed, the microstructural orientation of myelinated axons is maintained by oligodendroglial processes in the presence of experimentally induced dilatation of the brain extracellular space .
Successful distribution of vector within the white matter resulted in a brisk immune response characterized by a rapid infiltration of ED1 + activated microglia and CD8 + T cells. In the context of treating brain tumors, the induction of an anti-HSV CD8 response is likely to enhance the tumor cell kill and therefore treatment efficacy, but at the potential cost of promoting treatment toxicity. Although no animals in our study developed detectable neurological deficits, the long-term consequences of widespread distribution and cell transduction with HSV-1 vectors within the brain and the resultant vigorous immune response requires careful evaluation. This necessity is emphasized by the potential for significant vector escape from the relatively immune-privileged brain into the systemic circulation with the subsequent development of an adaptive immune response. Realistically, this could be mediated through tissue damage associated with cannula implantation, infusion-related damage, or drainage of vector through the perivascular spaces. Further research is critical for establishing whether an adaptive immune response does occur and, if it does, whether it leads to an enhanced antitumor response or leads to detrimental effects on normal brain.
The potential risks of viral vector leakage into the CSF and the development of an immune response emphasize the importance of visualizing vector distribution clinically. As such, a number of T 1 and T 2 contrast agents have been developed to act as surrogate markers of vector distribution . However, co-infusion of vector with a surrogate marker adds significant complexity and regulatory hurdles to the clinical administration of gene therapy vectors to the brain. Nevertheless, we have demonstrated that T 2 -weighted magnetic resonance imaging (MRI) is a feasible strategy for approximate visualization of infusate distribution, although sparse cell transduction clearly extended beyond the margins of this high signal. Finally, in view of the apparent perivascular and possible axonal transport of vector augmenting the distribution achieved directly by CED, it is highly unlikely that the use of surrogate markers or MRI would offer a realistic prediction of the final distribution of HSV vector-mediated transduced cells.
Related posts:
 Modified Oncolytic Herpesviruses for Gene Therapy of Cancer
Clinical Application of Plasmid-Based Cancer Vaccines
Modified Oncolytic Herpesviruses for Gene Therapy of Cancer
Clinical Application of Plasmid-Based Cancer Vaccines
 Imaging of Oncolytic Virus Gene Expression
Ethics in Translational Gene Transfer Research
Imaging of Oncolytic Virus Gene Expression
Ethics in Translational Gene Transfer Research
 Viral Insertion Site Detection and Analysis in Cancer Gene Therapy
Viral Insertion Site Detection and Analysis in Cancer Gene Therapy
 Selectively Replicating Oncolytic Adenoviruses Combined with Chemotherapy, Radiotherapy, or Molecular Targeted Therapy for Treatment of Human Cancers
Selectively Replicating Oncolytic Adenoviruses Combined with Chemotherapy, Radiotherapy, or Molecular Targeted Therapy for Treatment of Human Cancers
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


