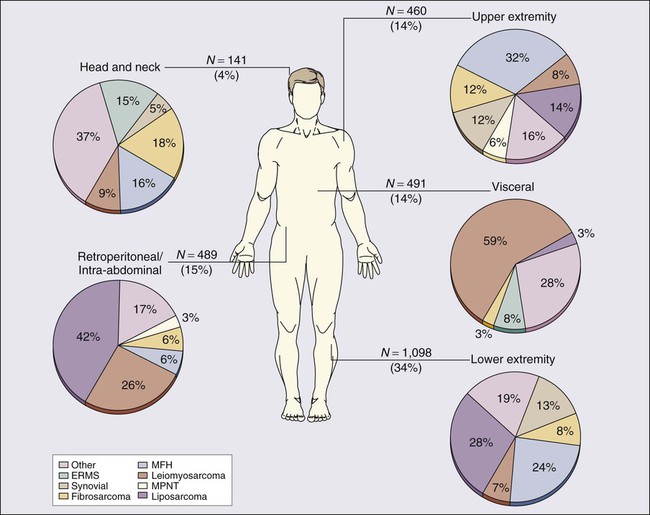
93 Lee J. Helman and Robert G. Maki • In the United States, there are 11,280 new cases annually; fewer than 1% of cancer diagnoses. • No specific etiologic agent is identifiable in the majority of cases. • Occasional cases are related to previous radiation, chemical exposure, alkylating chemotherapeutic agents, or chronic lymphedema. • Genetic conditions related to soft-tissue sarcoma include neurofibromatosis, tuberous sclerosis, basal cell nevus syndrome, Gardner syndrome, and Li-Fraumeni syndrome. Diagnosis and Evaluation of Extent of Disease • Core-needle biopsy (large lesions) or excisional biopsy (small lesions). • Pathological review of histologic subtype, grade, and assessment of margins (excisional biopsies). • Magnetic resonance imaging or computed tomography (CT) of primary site. • Chest x-ray for low-grade tumors and high-grade T1 lesions, chest CT for high-grade T2 tumors. • High-grade histology, deep location, and T2 tumor size are independent adverse prognostic factors for distant metastasis and survival. • Presentation with recurrent disease and positive surgical margin (gross or microscopic) are independent adverse prognostic factors for local recurrence. • Individual patient prognosis may be estimated by using a nomogram or newer techniques. • The American Joint Committee on Cancer (International Union Against Cancer) system uses criteria that include grade, size, and location relative to the investing muscular fascia, nodal status, and distant metastases. Other means to characterize risk of recurrence of soft-tissue sarcomas include nomograms and Bayesian belief networks. • Surgical resection with an adequate margin of normal tissue; for extremity lesions, a limb-sparing approach is possible in more than 90% of patients and offers survival comparable to amputation without the associated morbidity. • For most patients, local control is improved with preoperative or postoperative radiotherapy, with size and primary site also impacting its use. • The role of chemotherapy for high-risk patients remains controversial, but chemotherapy is used at several major centers for high-risk patients, especially for extremity tumors with known chemotherapy sensitivity, preoperatively when possible. • In addition to surgery, 3 years of adjuvant imatinib is the standard of care for high-risk gastrointestinal stromal tumor (GIST). • Local recurrence rates vary depending on the anatomic primary site and the adequacy of local therapy; for extremity lesions, approximately 20% of patients experience locally recurrent disease. • Systemic cytotoxic chemotherapy with selective use of surgery is the mainstay of therapy for patients with metastatic disease. • Kinase-directed therapy is clinically beneficial for patients with GIST; pazopanib was shown active in patients with other types of sarcoma who failed to respond to other systemic therapies, but has comparatively limited benefit. • For the small subset of patients who experience isolated (solitary) lung metastases, 20% to 50% 3-year survival rates have been reported with metastasis resection. Soft-tissue sarcomas (STSs) comprise a group of relatively rare, anatomically and histologically diverse neoplasms. Most STSs share a common embryologic origin, arising primarily from tissues derived from the mesodermal or ectodermal germ layers, distinct from carcinomas, which arise from the endodermal germ layer, although some sarcomas such as angiosarcomas also have an endodermal origin. Although the somatic soft tissues account for as much as 75% of total body weight, neoplasms of the soft tissues are comparatively rare, accounting for less than 1% of adult malignancies and 15% of pediatric malignancies. The relative rarity of these tumors, coupled with the histologic diversity of tumors, has led to studies that include diverse STS subtypes in a single study group, making it difficult to develop specific therapies for specific tumor types, especially in adult STSs. The annual incidence of STSs in the United States is about 11,280 new cases, comparable to the incidence of testicular cancer.1 However, an estimated 3900 patients die annually of STS—a rate nearly tenfold greater than is seen with testicular cancer—emphasizing the comparatively high overall mortality rate that is seen with this type of tumor. No specific etiologic agent may be identified in the majority of patients with STS. There are a number of recognized associations between environmental factors and the subsequent development of sarcoma; these are summarized in Table 93-1. The development of sarcoma has been reported after the use of ionizing radiation for the treatment of lymphoma2; solid tumors of the head and neck,3 breast,4,5 gynecologic organs, and skin; and benign conditions including endometriosis, tuberculous arthritis,6 and benign thymic enlargement. The vast majority of radiation-associated sarcomas are high-grade (87%), and the predominant histologies include undifferentiated pleomorphic sarcoma (formerly termed malignant fibrous histiocytoma), angiosarcoma, malignant peripheral nerve sheath tumor, osteosarcoma and others; translocation-associated sarcomas are rare, in comparison, and teleologically it is difficult to understand how random DNA damage leads to a specific chromosomal translocation.7–10 Table 93-1 Soft-Tissue Sarcoma: Predisposing Environmental Factors By criteria that were described initially by Cahan and colleagues,11 radiation-induced sarcomas appear no sooner than 4 years after the therapeutic radiation12,13 and often appear decades later. More recently, however, a shorter 2-year period after diagnosis has been used to define radiation-related disease, and the median time from radiation to development of a secondary sarcoma is 8 to 10 years. Contrary to early theories, recent studies have suggested that both orthovoltage and megavoltage treatments are sarcomagenic9,14 at doses from 8.8 to 70 Gy. In a carefully conducted case-control analysis, the Late Effects Study Group found 64 cases of osteosarcoma in 9170 patients who had survived more than 2 years after the diagnosis of a variety of cancers. A dose-response relationship was found between the radiation dose and the subsequent development of osteosarcoma, with a relative risk ranging from 0.6-fold in patients who received less than 10 Gy to 38.3-fold in those who received more than 60 Gy.15 A complete understanding of the underlying biology of radiation-induced malignancy remains elusive. Host-related factors (especially young age) and treatment (especially its intensity) both seem to play a role in a complex relationship.16 Underlying genetic susceptibility is also important and is governed by factors such as deletion or mutation of tumor suppressor and DNA repair genes. This scenario likely means that subgroups of the populations have a greater risk than was previously anticipated, whereas others are at lower risk than was believed. The identification of osteosarcoma as a common second malignancy following radiation therapy for retinoblastoma is an example of a high-risk scenario. It is probable that the greater intensity of multimodality treatments for cancer that have been introduced in recent years with the goal of improving cancer control and survival will result in an increase in the rate of radiation-induced malignancies. For example, the use of radiation after lumpectomy for ductal carcinoma in situ may result in unnecessary second cancers, which are avoidable in over 95% of patients with the use of mastectomy and reconstruction. The most common postradiation cancer appears to be sarcoma, especially considering its baseline incidence rates.16 Sarcomas have also been weakly associated with exposure to various chemical agents. A number of conflicting reports have emerged that suggest a relationship between occupational exposure to phenoxyacetic acids (found in some herbicides) and chlorophenols (found in some wood preservatives). Studies from Sweden demonstrated a link between phenoxy herbicide exposure in forestry workers and the subsequent development of sarcoma.17–20 However, additional investigations in the United States, New Zealand, and Finland have not confirmed this relationship.23–23 Similarly, there has been no demonstrable increase in the risk of sarcoma in Vietnam veterans exposed to Agent Orange (dioxin or TCDD [2,3,7,8-tetrachlorodibenzo-p-dioxin]).24 Hepatic angiosarcomas have been associated with exposure to a number of compounds, including Thorotrast (a colloidal suspension of thorium dioxide that was formerly used as an intravenous contrast agent in radiologic imaging procedures),27–27 vinyl chloride,30–30 and arsenic.31,32 Recent studies have suggested a relationship between exposure to alkylating chemotherapeutic agents and the subsequent development of sarcomas. Osteosarcomas have been reported after cyclophosphamide treatment for pediatric acute lymphoblastic leukemia.33,34 In the recent report from the Late Effects Study Group, prior chemotherapy, particularly with melphalan, procarbazine, nitrosoureas, or chlorambucil, was found to be an independent risk factor for the development of sarcoma.15 The relative risk of sarcoma increased with cumulative drug exposure. Chronic lymphedema may be a factor in the development of angiosarcoma. These neoplasms have been noted to arise in the chronically lymphedematous arms of women who were treated for breast cancer with radical mastectomy (Stewart-Treves syndrome).35,36 Lower-extremity angiosarcomas have also been observed in patients with congenital lymphedema or filariasis complicated by chronic lymphedema.37 A recent history of trauma is often elicited from sarcoma patients, particularly those with extremity sarcoma. Usually, the interval between the traumatic event and the diagnosis of sarcoma is short, making a causal relationship unlikely. Some reports have suggested, however, that chronic inflammatory processes may be a risk factor for sarcoma. Shrapnel, bullets, intramuscular iron injections, and foreign-body implants have been implicated.38 Germline mutations can play an important role in the development of STSs (Table 93-2). These genetic changes are identified with or similar to the genetic changes that are seen in corresponding sporadic sarcomas (Table 93-3). Mechanistically, the proteins encoded by the altered genes are involved in maintenance of the genome through DNA repair and the cell cycle. Table 93-2 Germline Mutations Associated with Soft-Tissue Sarcoma Table 93-3 Selected Molecular Alterations Identified in Sarcoma The epidemiological relationship between the development of STS and inherited syndromes associated with a predisposition to neoplasia (e.g., neurofibromatosis and Li-Fraumeni syndrome) has been appreciated for decades.39,40 For example, patients with neurofibromatosis have a 7% to 10% lifetime risk of developing a malignant peripheral nerve sheath tumor (MPNST).41 A sudden increase in the size of any neurofibroma suggests malignant transformation.42,43 The mechanisms underlying the transformation from a benign neurofibroma to MPNST are not well understood. However, loss-of-function mutations in the NF1 gene, which are found in patients with neurofibromatosis, result in activation of one of the ras signaling pathways, a well-known mechanism that has been identified in a variety of cancers.44 It has been observed that secondary MPNSTs (arising from a prior neurofibroma) have deletions of 17p (particularly 17p12–17p13.1) and mutations at the region of the TP53 tumor suppressor gene.47–47 Thus it is postulated that an initial alteration in the NF1 gene contributes to the formation of a benign neurofibroma through activation of the ras pathway and that secondary mutations in the TP53 gene allow the transformation into MPNST. Because mutations in TP53 lead to an inability to control DNA damage via the cell cycle, they also enable rapid accumulation of other mutations, which encode mutant proteins that undoubtedly play an important role in sarcomagenesis. Li-Fraumeni syndrome was identified when relatives of pediatric STS patients were noted to have an increased frequency of diverse and often multiple primary cancers.39,48 The neoplasms that were noted in relatives included some STSs, premenopausal breast cancers, brain tumors, adrenocortical carcinomas, leukemias, and occasional germ cell tumors.49 Follow-up of Li-Fraumeni kindreds over 2 decades has revealed that the majority of individuals have cancer at young ages, with 79% of those affected younger than age 45 years at the time of diagnosis of malignancy.50 The observed cancer distribution in families is believed to fit a rare autosomal dominant mode of genetic transmission with high penetrance.51 Recent molecular genetic studies have identified germline TP53 mutations in the majority of patients with Li-Fraumeni syndrome.52 Germline mutations in CHK2, another component of the cell-cycle checkpoint machinery encoded by a gene located on 22q11, are responsible for another subgroup of patients with Li-Fraumeni syndrome, and other Li-Fraumeni–like patients’ kindreds without TP53 alteration have been described.53 Pediatric patients with familial retinoblastoma have a 13q chromosomal deletion54 and an increased incidence of osteosarcoma and other neoplasms, including STS.15,55,56 The retinoblastoma (Rb1) protein is expressed ubiquitously in normal cells and is a well-known tumor suppressor, involved in maintaining the integrity of the genome through control of the cell cycle. Interestingly, not only is the Rb1 gene that is mutated in osteosarcomas associated with retinoblastoma, but abnormality or absence of the Rb1 gene product has also been observed in multiple other malignancies, including sporadic osteosarcomas, breast cancer,57 small cell lung cancer,58 and STSs.59 Familial adenomatous polyposis, a subset of which includes Gardner syndrome with its desmoid tumors, is caused by germline mutations in the APC gene.62–62 Patients with Gardner syndrome experience desmoid fibromatosis at a younger age than do patients with sporadic desmoids.63 With the use of prophylactic colectomy, progressive mesenteric desmoid tumor is now a significant cause of death of patients who have familial adenomatous polyposis,64 as are second neoplasms such as ampullary and duodenal cancers. The APC gene is involved in the Wnt cell-signaling pathway. One of the normal functions of the APC protein is to bind β-catenin.65 Thus loss-of-function mutations of APC result in the activation of transcription of oncogenes by β-catenin. Consistent with the importance of this pathway in desmoid tumors, APC loss and CTNNB1 (encoding β-catenin) mutations are also identified in sporadic desmoid fibromatosis.68–68 Rhabdoid predisposition syndrome is due to inactivating germline mutations in the INI1 gene.69 Patients with this syndrome experience one or more extrarenal and/or renal rhabdoid tumors. INI1 is a member of the SWI/SNF protein complex, which controls gene expression globally through its ability to alter chromatin structure.70 Thus loss of INI1 gene expression results in other changes in gene expression, specifically activation of oncogenes. Rhabdoid tumors have loss-of-function mutations in both copies of the INI1 gene.71 Werner syndrome is a rare genetic instability syndrome caused by mutations in the WRN gene.72,73 Affected patients age prematurely and are at greatly increased risk for a variety of cancers, including STSs. The WRN gene encodes a protein involved in DNA repair74 and loss of WRN protein function leads to genetic instability, accumulation of genetic mutations, and, ultimately, predisposition to rapid aging and cancer. Germline mutations in the KIT oncogene are found in patients with familial gastrointestinal stromal tumor (GIST) syndrome.75,76 Activating KIT or PDGFRA mutations are also identified in over 90% of sporadic gastrointestinal stromal tumors (GISTs).77 Patients with the familial syndrome experience, to varying degrees, skin hyperpigmentation, urticaria pigmentosa, and cutaneous mast cell disease in addition to one or more GISTs.78 Activating KIT mutations have been shown to lead to ligand-independent activation of the KIT receptor tyrosine kinase pathway, which results in dysregulated cell growth, and are thought to be the first step in the pathogenesis of GISTs.75 Interestingly, the identification of the important role of KIT in the pathogenesis of GISTs has led to treatment with imatinib,79 the oral multitargeted receptor tyrosine kinase inhibitor (see “Prognostic Factors as Therapeutic Targets” and “Gastrointestinal Stromal Tumors” below). A large number of sarcomas have been found to have consistent chromosomal abnormalities (see Table 93-3).80 These chromosomal rearrangements are important diagnostically, may be important prognostically (see “Potential Molecular Prognostic Factors” below), and have shed light on the pathogenesis of sarcomas. Some of the specific translocations, such as those seen in dermatofibrosarcoma protuberans or tenosynovial giant cell tumor provide targets for pharmacologic therapy (see the section on treatment of GISTs). Benign soft-tissue neoplasms also harbor chromosomal rearrangements. Chromosomal translocations are the most common cytogenetic abnormality in soft-tissue neoplasms and are likely responsible for the initiation of tumorigenesis in most cases.80 Deletions and trisomies have also been reported and are thought to represent secondary changes involved in tumor progression. Deletions tend to represent loss of tumor suppressor genes, whereas trisomies suggest the presence of an oncogene. Although we know much about the principal tumorigenic events in many sarcomas, defining the secondary changes has been much more problematic and is an area of intense study with the advent of large-scale tumor DNA and RNA sequencing and related genomic techniques. Cloning and molecular analysis of the various genetic aberrations that characterize different sarcomas have revealed the different pathogenetic mechanisms that underlie these tumors. Translocations typically create chimeric transcription factors or growth factors that result in deregulation of transcription or growth control. A typical example of a chimeric transcription factor is the PAX3-FOXO1 fusion protein, which has been shown to activate a complex myogenic transcriptional program when the protein is expressed in a fibroblast cell line.81 Infantile fibrosarcoma is characterized by a translocation involving chromosomes 12 and 15 that encodes a chimeric ETV6-NTRK3 constitutively activated growth factor receptor, which remarkably is found in other cancers such as secretory breast cancer, a subset of acute myelogenous leukemia, and a form of salivary gland carcinoma.82 Other oncogenic proteins appear to act by a mechanism that remodels chromatin structure, which is known to have a profound influence on gene expression (e.g., INI1 mutations in rhabdoid tumors).71 Specific chromosomal rearrangements are very useful in the diagnosis of STSs. Beyond the obvious benefit of providing further objective proof of a diagnosis in morphologically typical cases, the detection of chromosomal aberrations may facilitate the diagnosis of lesions that are difficult to characterize by standard histopathological, ultrastructural, and immunohistochemical techniques.83,84 For example, the presence of the translocation t(X;18)(p11;q11) has been used to confirm the diagnosis of synovial sarcoma in poorly differentiated cases that were diagnostically very challenging.87–87 Similarly, the finding of the characteristic translocation t(11;22)(q24;q12) in a small round blue cell tumor supports the diagnosis of Ewing sarcoma/primitive neuroectodermal tumor (PNET).88 By the same token, a morphologic sarcoma subtype without an expected translocation may suggest a variant translocation, as has been observed in Ewing sarcoma–like small round blue cell tumors that lack the EWSR1-FLI1 translocation but rather contain CIC-DUX4 or a pericentric X chromosome inversion.91–91 Translocations may be identified by a variety of genetic analyses, spanning older techniques such as cytogenetic analysis to fluorescence in situ hybridization, reverse transcriptase polymerase chain reaction, and RNA sequencing. A detailed description of these techniques is beyond the scope of this chapter, but each technique has its advantages and disadvantages. Cytogenetic analysis requires fresh (living) tissue, because the cells need to be cultured before karyotypic analysis, and it largely has been supplanted by newer techniques. Fluorescence in situ hybridization does not require fresh or frozen tissue and frequently may be used with paraffin-embedded material; it is a standard diagnostic tool in many pathology laboratories. It is noted that evidence of a translocation by fluorescence in situ hybridization is not absolutely diagnostic of a sarcoma subtype, because one gene, such as EWSR1, may be involved in many different types of sarcomas, and such data must be incorporated with other data regarding the tumor sample. Reverse transcriptase polymerase chain reaction is a more sensitive technique that may be performed on fresh, frozen, or paraffin-embedded tissues. The major drawback of reverse transcriptase polymerase chain reaction is the relatively high false-positive rate, which results from its sensitivity. Meticulous care is required to prevent problems from contamination. Sequencing of the RNA complement of the genome allows for detection of mutations, translocations, and, to some degree, gene amplification, and is expected to be used more frequently over time.92 Many sarcomas are characterized by several different translocations, most of which are mutually exclusive (see Table 93-3). For instance, alveolar rhabdomyosarcoma is characterized by a translocation involving chromosomes 2 and 13, which results in fusion of the PAX3 and FOXO1 genes, or by a translocation involving chromosomes 1 and 13, which results in fusion of the PAX7 and FOXO1 genes.93–96 Sarcomas within a subtype may also have differences in the specific exons that are involved in each of these different translocations. It has been proposed that this heterogeneity may result in differences in prognosis (see “Potential Molecular Prognostic Factors” below). For example, Ewing sarcoma/PNET and synovial sarcoma possess genetic variations that have been suggested to have prognostic significance, further underscored by the Ewing-like sarcomas that lack EWSR1 fusions but contain other translocations.89,91,97–102 Future research will hopefully establish whether cytogenetic and molecular factors may be used as a basis for therapeutic decisions and the prediction and evaluation of response to treatment. The identification of genetic alterations with high specificity for different sarcomas will also identify specific therapeutic targets. This has already resulted in the successful treatment of a variety of sarcomas, for example, GISTs, dermatofibrosarcoma protuberans (DFSP), and tenosynovial giant cell tumor/pigmented villonodular synovitis (TGCT/PVNS), and to a lesser degree, angiosarcoma, desmoid tumor, and Ewing sarcoma.79,103–105 Approximately 90% of GISTs harbor activating mutations in the KIT oncogene, which result in ligand-independent activation of the KIT receptor tyrosine kinase pathway.77 Imatinib, a small molecule drug that is administered orally and inhibits the KIT receptor tyrosine kinase, is highly efficacious in the treatment of GIST (see section of treatment of GISTs). DFSP is characterized by translocations involving the COL1A1 and PDGFB genes, which result in activation of the platelet-derived growth factor-β (PDGF-β) pathway in what is believe to be an autocrine fashion. Imatinib is active against the PDGF-β pathway and has been shown to be effective in the treatment of a small number of DFSPs. In a similar way, TGCT/PVNS with its t(1;2) involving COL6A3-CSF1 is responsive to imatinib by virtue of its inhibition of the CSF1 receptor.106 It is noteworthy to mention two common benign soft-tissue tumors—leiomyomas and lipomas—that have a high frequency of chromosomal rearrangements of chromosome 12q that involves the high-mobility protein group gene HMGIC/HMGA2.107,108 These translocations are not seen in the corresponding leiomyosarcomas or liposarcomas, indicating that in these tumors, the benign form is not a precursor of the malignant counterpart. Other benign soft-tissue neoplasms contain specific translocations, such as nodular fasciitis [t(17;22)(p13;q13) MYH9-USP6] or angiomatoid fibrous histiocytoma [t(12;22)(q13;q12) EWSR1-ATF1], underscoring the idea that translocations can occur in tumors anywhere in the spectrum of connective tissue tumors from benign to malignant.109,110 A second major subset of STSs is characterized by aneuploidy and the lack of specific fusion genes. This group of sarcomas includes such tumors as leiomyosarcomas, MPNST, and fibrosarcomas (see Tables 93-3 and 93-4). These tumors tend to occur in older patients and appear to have a relatively high frequency of mutations in the p53 and retinoblastoma (Rb) signaling pathways.111,112 These tumors are characterized by chromosomal gains and losses that presumably target tumor suppressor genes (in the event of losses) and oncogenes (in the event of gains). A special case appears to be well-differentiated/dedifferentiated liposarcoma, with amplifications of cell-cycle regulatory genes CDK4 and HDM2 and others on chromosome 12q. The cooperation of these genes to create a unique type of aneuploid sarcoma remains a current challenge in the field. Supporting the clinical data from syndromic sarcomas, alterations disrupting chromosomal mechanics and DNA repair appear to be involved in sarcomagenesis. Table 93-4 Sarcomas with Complex Karyotypes Prospectively collected databases of STS patients, some of which contain data on more than 10,000 patients, demonstrate that STSs are described in essentially all anatomic sites. Figure 93-1 outlines the anatomic sites and site-specific histologic subtypes of 4207 sarcomas treated at a single referral institution. Approximately half of all STSs occur in the extremities (lower: 34%; upper: 14%), where the most common histopathological subtypes are well-differentiated, myxoid, and round cell liposarcoma (28%) and undifferentiated pleomorphic sarcoma (UPS, formerly termed malignant fibrous histiocytoma) (24%). Retroperitoneal sarcomas make up 15% of all STSs, well-differentiated or dedifferentiated liposarcoma being the predominant histologic subtype (42%). Visceral sarcomas make up an additional 14%, whereas the head and neck sarcomas make up approximately 4%. An alternative way to index STSs is by their differentiation. Tumors may be grouped broadly into adipocytic tumors, fibroblastic/myofibroblastic tumors, so-called fibrohistiocytic tumors, smooth muscle tumors, pericytic (perivascular) tumors, PNETs, skeletal muscle tumors, vascular tumors, osseous tumors, and tumors of uncertain differentiation (Table 93-5).113 Classification is based on clinical, histologic, ultrastructural, immunohistochemical, and genetic features. Electron microscopic evidence of cellular substructures, neurofibrils, microfilaments, actin-myosin complexes, and dense bodies, has historically been used to define the tissue of origin.114 However, the widespread availability of commercial antibodies for immunohistochemical analysis has all but eliminated the need for electron microscopic examination. Immunohistochemical staining for proteins that are characteristic of smooth muscle (smooth muscle actin and desmin), skeletal muscle (muscle-specific actin, desmin, and myogenin), blood vessels (factor VIII, CD34, and CD31), and epithelial tissue (epithelial membrane antigen and cytokeratins) often facilitates reliable classification, with genetic proof of a sarcoma translocation considered to be iron-clad evidence of a specific sarcoma subtype.115,116 However, because the identical translocation may be found in more than one type of cancer, such as the ASPL-TFE3 translocation of both alveolar soft part sarcoma and papillary renal cancer, translocation data cannot be used in isolation. Table 93-5 Histologic Classification of Soft-Tissue Sarcoma The tissue of origin classification scheme is the basis for the 2013 World Health Organization classification system for sarcomas.113,117 The World Health Organization classification system is reproducible for most sarcomas. As the degree of histologic differentiation declines, however, the determination of the tissue of origin becomes increasingly difficult. In particular, despite advanced immunohistochemical and molecular analyses, determining the tissue of origin for some soft-tissue tumors may be difficult, occasionally arbitrary, and sometimes impossible. This leads to significant disparities in diagnoses among pathologists. Discrepancies between the original histologic diagnosis and the subsequent diagnosis by an expert reviewer have been noted in as many as 25% of cases, although these data predate modern immunohistochemical and genetic analyses.118,119 Review of suspicious tissue specimens at an expert center is therefore wise, because the degree of expertise in correctly diagnosing some of the 50 or more types of rare and unusual sarcoma is directly related to the number of sarcomas that a pathologist has seen, and, increasingly, the specific molecular diagnostic tools needed to identify rare sarcoma subtypes. It is important to classify STSs as precisely as possible because of major differences in their clinical behavior and in their susceptibility to different therapies. For example, a few STSs, including epithelioid sarcoma, clear cell sarcoma, angiosarcoma, and rhabdomyosarcoma have a greater risk of regional lymph node metastasis.120,121 In one single-institution study, the overall rate of nodal metastasis at the time of sarcoma presentation was under 3%; however, the rate was much higher for angiosarcoma (13%), embryonal rhabdomyosarcoma (14%), and epithelioid sarcoma (17%).120,121 Patterns of distant metastases also differ for subtypes of sarcoma. For example, myxoid/round cell liposarcoma tends to metastasize to soft-tissue sites, including the retroperitoneum as well as to the spine and pelvis and other bone marrow sites,122,123 and patients with myxoid/round cell liposarcoma often present with metastatic disease. If a myxoid/round cell liposarcoma is identified in the abdomen, the thighs should be examined for an occult primary tumor. The majority of what had been suspected to be primary myxoid liposarcomas of the abdomen and retroperitoneum are actually misdiagnosed well differentiated / dedifferentiated liposarcomas that have myxoid features (and usually metastasize far less often than myxoid/round cell liposarcoma).124 Histologic classification alone does not always provide enough information to predict the clinical behavior of STSs. For many sarcomas, histologic grading provides additional information that can aid in predicting biological behavior and planning treatment. The spectrum of grades varies among specific histologic subtypes (Fig. 93-2). For example, liposarcomas exhibit wide variations in grade, whereas Ewing sarcomas/PNETs are always considered high-grade. In careful comparative multivariate analyses, histologic grade has been the most important prognostic factor in assessing the risk for distant metastasis and tumor-related mortality.127–127 Several grading systems have been proposed, but there is no consensus regarding the specific morphologic criteria that should be employed in the grading of STSs. The two most important criteria appear to be the mitotic index and the extent of tumor necrosis.
Sarcomas of Soft Tissue*
Introduction
Etiology and Epidemiology
Environmental Factors
Factor
Agent
Patient Population
Comment
Radiotherapy
Orthovoltage and megavoltage radiation
Therapeutic radiation patients
Most commonly undifferentiated pleomorphic sarcoma and osteosarcoma; dose-response relationship
Chemotherapy
Alkylating agents: cyclophosphamide, melphalan, procarbazine, nitrosourea, and chlorambucil
Pediatric cancer patients
Relative risk of bone sarcoma increased with cumulative drug exposure
Chemical exposure
Phenoxyacetic acids: 2,4-dichlorophenoxyacetic acid (2,4-D); 2,4,5 trichlorophenoxy acetic acid (2,4,5-T); 2 methyl-4 chlorophenoxyacetic acid (MCPA)
Forestry and agricultural workers
Phenoxy herbicide and defoliant exposure
2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD); dioxin
Vietnam veterans
No clear link demonstrable for dioxin (Agent Orange)
Chlorophenols
Sawmill workers
Thorotrast
Diagnostic x-ray patients
Hepatic angiosarcoma
Vinyl chloride
Vinyl chloride workers
Hepatic angiosarcoma
Arsenic
Vineyard workers
Hepatic angiosarcoma after exposure to arsenical herbicides
Chronic lymphedema
Postsurgery patients
Postradiation patients
Patients with congenital lymphedema or filariasis
Stewart-Treves syndrome (lymphangiosarcoma)
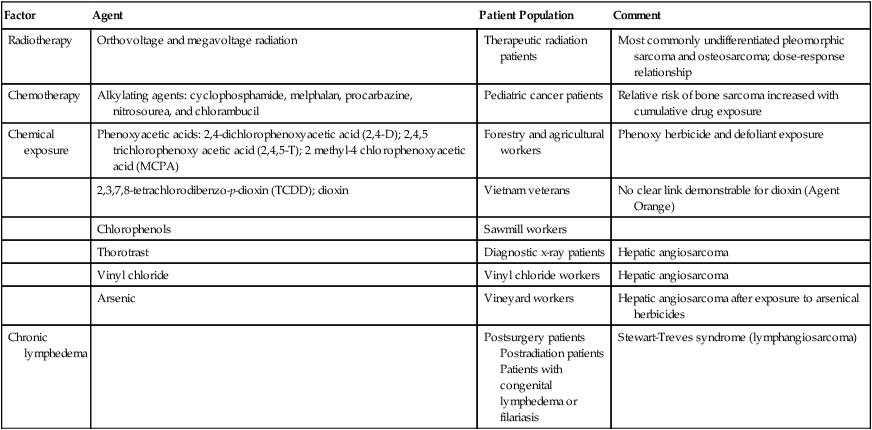
Genetic Predisposition
Syndrome
Inheritance Pattern
Locus
Gene
Associated Soft-Tissue Sarcomas
Familial gastrointestinal stromal tumor syndrome
AD
4q12
KIT
Gastrointestinal stromal tumor
Familial desmoid fibromatosis (Gardner syndrome)
AD
5q21
APC
Desmoid fibromatosis
Li-Fraumeni syndrome
AD
17p13,22q11
TP53, CHK2
Multiple types
Neurofibromatosis type I (von Recklinghausen disease)
AD
17q11
NF1
Malignant peripheral nerve sheath tumors
Retinoblastoma
AD
13q14
RB1
Multiple types
Rhabdoid predisposition syndrome
AD
22q11
SNF5/INII
Malignant rhabdoid tumors
Werner syndrome
AR
8p11–12
WRN
Multiple types
Tumor Type
Characteristic Cytogenetic Events
Molecular Events
Frequency (%)
Diagnostic Utility?
Alveolar soft part sarcoma
t(X;17)(p 11;q25)
ASPL-TFE3 fusion
>90
Yes
Extraskeletal myxoid chondrosarcoma
t(9;22)(q22;q12)
EWSR1-NR4A3 fusion
>75
Yes
Clear cell sarcoma
t(12;22)(q13;q12)
EWSR1-ATF1 fusion
>75
Yes
Desmoplastic small round cell tumor
t(11;22)(q13;q12)
EWSR1-WT1 fusion
>75
Yes
Dermatofibrosarcoma protuberans
Ring form of chromosomes 17 and 22
COLIA1-PDGFB fusion
>75
Yes
t(17;22)(q21;q13)
COLIA1-PDGFB fusion
10
Yes
Ewing sarcoma/peripheral primitive neuroectodermal tumor
t(11;12)(q24;q12)
EWSR1-FLII fusion
>80
Yes
t(21;22)(q12;q12)
EWSR1-ERG fusion
5-10
Yes
t(2;22)(q33;q12)
EWSR1-FEV fusion
<5
Yes
Fibrosarcoma, infantile
t(12;15)(q13;q26)
ETV6-NTRK3 fusion
>75
Yes
Trisomies 8,11,17 and 20
>75
Yes
Gastrointestinal stromal tumor
Monosomies 14 and 22
>75
Yes
Deletion of 1p
KIT or PDGFRA mutation
>25
>90
No
Yes
Inflammatory myofibroblastic tumor
2p23 rearrangement
ALK fusion genes
50
Yes
Leiomyosarcoma
Deletion of 1p
>50
No
Liposarcoma
Well-differentiated
Ring form involving chromosome 12q
>75
Yes
Myxoid/round cell
t(12;16)(q13;p11)
FUS-DDIT3 fusion
>75
Yes
Pleomorphic
Complex
EWSR1-DDIT3 fusion
<5
Yes
Undifferentiated pleomorphic sarcoma
Complex
>90
No
Malignant peripheral nerve sheath tumor
Complex; NF1 loss
>90
No
Rhabdoid tumor
Deletion of 22q
INI1 inactivation
>90
Yes
Rhabdomyosarcoma
Alveolar
t(2;13)(q35;q14)
PAX3-FOXO1 fusion
>75
Yes
t(1;13)(p26;q14), double minutes
PAX7-FOXO1 fusion
10-20
Yes
Embryonal
Trisomies 2q, 8, and 20
>75
Yes
Loss of heterozygosity at 11p15
>75
Yes
Synovial sarcoma
Monophasic
t(X;18)(p11;q11)
SS18-SSX1 or SS18-
>90
Yes
Biphasic
t(X;18)(p11;q11)
SSX2 fusion
SS18-SSX1 fusion
>90
Yes
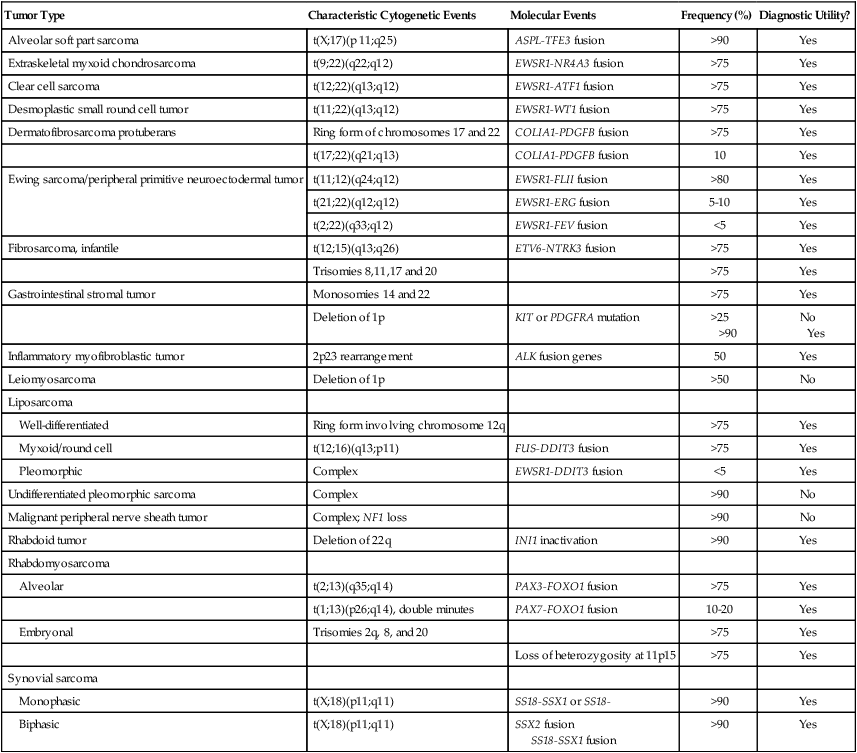
Chromosomal Rearrangements
Sarcomas with Complex Karyotypes
Type of Sarcoma
Resembles
Fibrosarcoma (other than congenital)
Fibrous tissue
Leiomyosarcoma
Smooth muscle
Undifferentiated pleomorphic sarcoma
Poorly differentiated
Osteosarcoma
Bone
Chondrosarcoma (types other than extraskeletal myxoid)
Cartilage
Liposarcoma (types other than myxoid)
Fat
Embryonal rhabdomyosarcoma
Skeletal muscle
Malignant peripheral nerve sheath tumor*
Nerve sheath
Angiosarcoma
Endothelium
Pathology
Classification
ADIPOCYTIC SARCOMAS
FIBROBLASTIC AND MYOFIBROBLASTIC SARCOMAS
SO-CALLED FIBROHISTIOCYTIC SARCOMAS
SMOOTH MUSCLE SARCOMAS
SKELETAL MUSCLE SARCOMAS
VASCULAR SARCOMAS
OSSEOUS SARCOMAS
SARCOMAS OF UNCERTAIN DIFFERENTIATION
NOTOCHORD-DERIVED TUMOR BEHAVING LIKE SARCOMAS

Histologic Grading
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Sarcomas of Soft Tissue