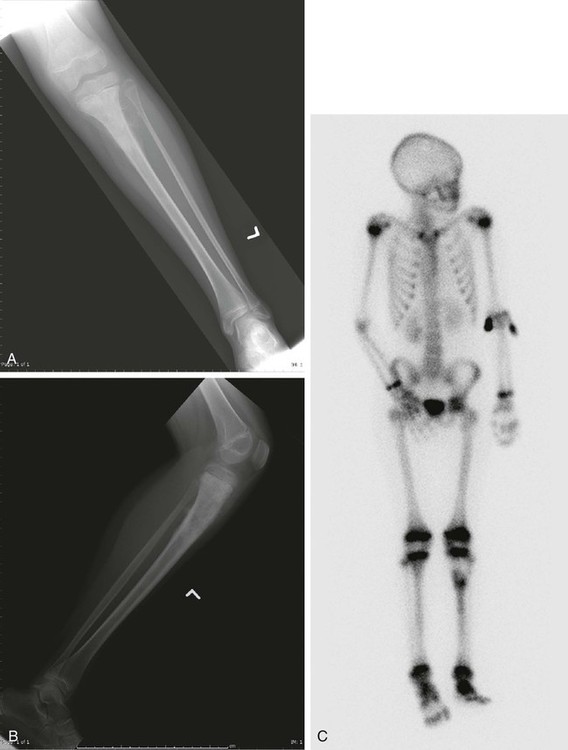
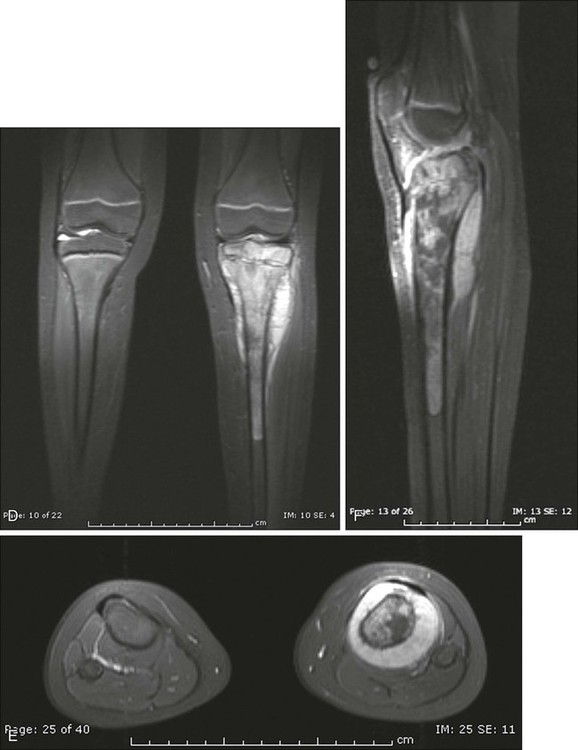
92 Megan E. Anderson, R. Lor Randall, Dempsey S. Springfield and Mark C. Gebhardt • More than 2800 new cases of bone sarcoma are diagnosed annually in the United States. • No specific etiologic agents are identified in the majority of cases. • Secondary neoplasms are related to known oncogenic factors. (e.g., ionizing radiation, alkylating chemotherapy agents, combinations of both). • Hereditary cancer syndromes (tumor suppressor genes) are responsible for some cases. Diagnosis and Radiographic Staging • Plain radiographs are recommended. • Magnetic resonance imaging (MRI) scan of primary tumor is the best radiographic study to obtain. • Chest computed tomography (CT) is indicated to evaluate for pulmonary metastases. • Whole-body technetium-99m (99mTc) bone scan is indicated to evaluate for metastases to other bones. • Positron emission tomography (PET) scanning is controversial and has yet to be generally accepted. • Needle or open biopsy is necessary for a tissue-specific diagnosis and to determine histologic grade. • In the pathology review, immunohistochemistry and cytogenetics are important. • Electron microscopic tissue occasionally is required. • Metastasis at presentation is a worse prognostic finding. • Histologic grade is the next most significant prognostic indicator. • Size is less significant, but lesions larger than 10 cm in diameter have a poor prognosis. • Tumor response to neoadjuvant chemotherapy is important in osteosarcoma and Ewing sarcoma. • Surgical margins of resection (minimum of a “wide” margin). • The American Joint Committee on Cancer now monitors grade (high and low), and size (8 cm); designates “skip” lesions (T3); and separates metastasis to bone from other sites (Mla, Mlb). • The Musculoskeletal Tumor Society monitors location. (intracompartmental and extracompartmental), grade (high and low), and metastasis (“skip” lesions, nodal, bone, and lung are all lumped together). • A wide surgical margin is recommended. • Limb-sparing procedures are appropriate for 70% to 90% of patients. • Adjuvant irradiation is not routinely used for bone sarcomas. • Local recurrence rates for limb-sparing procedures approach 5% or less. • Reconstruction methods can be tailored to patients’ needs. • New and improved biocompatible implant materials and improved designs are available. • The search continues for new drugs, drug schedules, potentiating agents, and improved dose intensity. • Identification of risk factors (e.g., cytogenetic, molecular genetic, and signal transduction abnormalities) will improve to identify new methods of potential treatment. It is estimated that 2890 new malignant tumors of bone (excluding multiple myeloma) were diagnosed in 2012 in the United States (http://seer.cancer.gov). The femur is the most common site, but primary sarcoma can occur in any bone. Osteosarcoma, Ewing sarcoma, and chondrosarcoma account for approximately 90% of all primary sarcomas of bone. The management of osteosarcoma and Ewing sarcoma includes chemotherapy and surgery, whereas chondrosarcoma is treated by surgery alone.1 The management of these patients, from initial evaluation and biopsy through surgical therapy and long-term follow-up, is labor intensive and technically demanding. Patients with a bone sarcoma should be treated in a center that has expertise in the management of these tumors. Currently, the staging system adopted by the Musculoskeletal Tumor Society (MSTS) in 1980 and modified in 1986 is accepted by most musculoskeletal oncologists.4–4 Malignant tumors are divided into only two histologic grades: low-grade malignant (G1) and high-grade malignant (G2). Low-grade malignant lesions (G1), comprising Broder I and II lesions, have a low probability of metastasis (25%). The majority of these tumors can be managed by relatively conservative surgical procedures and do not require chemotherapy. High-grade lesions (G2), Broder III and IV tumors, have a significantly higher incidence of metastases and require more radical surgical procedures and possibly neoadjuvant and/or adjuvant chemotherapy. Table 92-1 is a representative grouping of both low- and high-grade malignant tumors of bone and soft-tissue origin. Table 92-1 Radical margins of a bone sarcoma are achieved when the entire bone is removed. This process usually requires an amputation. A radical margin is rarely necessary.5 The American Joint Committee on Cancer (7th edition) has adapted the TNM (tumor–nodes–metastases) staging system to bone. The topography (T) of the primary tumor now includes size based on relevant published reviews, in which the greatest dimension (8 cm) has replaced the compartment concept. Also, T3 has now been assigned to patients with “skip” metastases (Table 92-2). Table 92-2 Definition of TNM Primary Tumor (T) Adapted from Edge SB, Byrd DR, Compton CC, et al., editors. AJCC cancer staging manual. 7th ed. New York: Springer Verlag; 2010. The problem of defining histopathological grade (G) has been addressed and now essentially consists of low- and high-grade lesions (Table 92-3). G1 and G2 have been combined into low-grade and G3 and G4 into high-grade histopathology. Currently, all Ewing tumors are classified as G4 or high-grade. This grouping is now identical to the G1 and G2 of the MSTS staging system. Table 92-4 shows the stage groupings. Here, the committee has appropriately addressed the difference in prognosis of patients who have sustained metastases to lung (Mla) and to other sites, including bone (Mlb). Table 92-3 *Ewing sarcoma is classified as G4. Adapted from Edge SB, Byrd DR, Compton CC, et al., editors. AJCC cancer staging manual. 7th ed. New York: Springer Verlag; 2010. Table 92-4 Adapted from Edge SB, Byrd DR, Compton CC, et al., editors. AJCC cancer staging manual. 7th ed. New York: Springer Verlag; 2010. Conventional bone radiography remains the single most useful initial study for bone tumor evaluation. The study should include anteroposterior and lateral projections of the lesion and ideally the whole bone in which the lesion is present. Malignant neoplasms usually result in ill-defined or “poorly marginated” radiographic margins with little or no reactive bone, loss of medullary trabeculation, and endosteal cortical erosion at the tumor–host bone interface, suggesting an active and destructive process. The pathological process biologically overwhelms the normal time-dependent reactive processes of bone formation. Therefore, the radiographic presence or absence of a reactive rim of bone is often useful in predicting the biological aggressiveness of the pathological process (Table 92-5). Neoplastic bone formation is often seen in osteosarcoma, and calcification is often seen in chondrosarcoma. Table 92-5 Radiographic Techniques Available Computed tomography (CT) is superior to magnetic resonance imaging (MRI) only for evaluating a small lesion in the cortex, subtle bone formation, or calcification; otherwise, MRI is the study of choice. CT remains the standard for evaluation of the chest for occult metastases. The cross-sectional display usually provides sufficient resolution (<0.5 cm) to demonstrate subpleural metastases long before they become evident on plain chest films. Before definitive therapy or local management of a potentially malignant lesion, a staging CT evaluation of the chest and mediastinum should be performed.6 Currently, MRI of the primary tumor appears somewhat predictive of tumor response to neoadjuvant therapy. Changes in the T2-weighted image signal intensity correlate with an obvious reduction in tumor volume (especially in Ewing sarcoma) and appear predictive of tumor necrosis.7 The addition of contrast enhancement does not appear to provide more viable tumor/necrotic tumor contrast than do T2-weighted images; however, the absence of contrast enhancement appears to be an indicator of tumor necrosis.7–10 Positron emission tomography (PET) has been used to predict response to chemotherapy for Ewing sarcoma and osteosarcoma. PET scan might be valuable in the initial screening, but this has yet to be determined.11,12 The staging biopsy might well be the most important and difficult procedure that is performed in the patient’s management (Box 92-1). The placement, length, and orientation of the biopsy scar and the anatomic compartments that are contaminated during the biopsy procedure dictate which tissues and how many surgical compartments will require removal for local tumor management and limb-sparing surgery. A thorough knowledge of the soft-tissue anatomic planes and muscle compartments is mandatory before proceeding with bone biopsy. Consideration should be given to the location and type of biopsy to be used, whether fine-needle aspiration biopsy, core-needle biopsy, or open biopsy. In addition, it is critical that adequate diagnostic tissue be obtained so that an accurate diagnosis can be made. Usually, a core-needle biopsy is sufficient. High-grade osteosarcoma used to be a fatal neoplasm leading to metastases and death of 90% of patients despite aggressive local control, including radical amputations and/or radiotherapy. Previous versions of this chapter outlined the evolution of the use of adjuvant chemotherapy and the dramatic improvement in survival and event-free survival (EFS) that followed. Initially, the benefits of adjuvant chemotherapy were questioned, leading to the need for a randomized study comparing surgical management alone with surgery followed by multiagent chemotherapy. The active drugs were shown to be doxorubicin, high-dose methotrexate, and cisplatin. A randomized study, including both the randomly assigned patients and those who chose whether or not to have adjuvant chemotherapy, clearly demonstrated the benefits of adjuvant chemotherapy.13,14 Surgical advances paralleled the advances in medical management of osteosarcoma patients. Originally, disarticulation or resection of the entire involved bone was recommended for surgical management of osteosarcoma.15 This recommendation was partly a result of the intramedullary origin of the tumor with proximal intramedullary growth and the reported 25% incidence of intramedullary “skip” metastases.16 Later studies reviewing the local recurrence rates for patients whose primary management was transmedullary amputation alone revealed local recurrences in approximately 5% to 10%, suggesting that the incidence of “skip” or intraosseous metastasis was probably lower than originally believed. The general standard of surgical management of patients with extremity osteosarcoma in 1980 included transmedullary amputation approximately 5 to 7 cm proximal to the intramedullary extent of the tumor.17 One event that led to the popularization of limb salvage was the use of preoperative chemotherapy. Initially implemented during the time it would take to manufacture a custom prosthesis, it became apparent that the tumor showed clinical and radiographic “response” to the preoperative (neoadjuvant) chemotherapy. This seemed to make the subsequent surgery easier, if not safer, and gave the surgeon 10 to 12 weeks to work with the patient to decide on the best surgical option. There was initial concern that the delay in the resection might worsen the prognosis, but this issue was addressed in a randomized trial that showed no apparent advantage to having the surgery initially compared to the neoadjuvant mode of administration.18 In addition, it was learned that the histologic response to the preoperative chemotherapy was a predictor of outcome and, second only to the presence of metastases at diagnosis, was the best predictor of survival.18,19 Conventional, or classic, osteosarcoma makes up the majority of all osteosarcomas. It occurs primarily in the metaphyses of adolescents with open physes or in young adults. Most patients with classic osteosarcoma are younger than age 30 years, and many have no apparent predisposing factors.20 The lesion most often arises in the larger, more active metaphyses (e.g., distal femur, proximal tibia, proximal humerus), but also can arise in the flat bones of the pelvis, skull, scapula, and ribs, and in the spine. Overall, the majority of the lesions develop in the extremities and pelvis.21 An epidemiological study conducted in Sweden between 1971 and 1984 investigated possible changes in the typical features of 227 conventional osteosarcomas. The mean annual incidence was 2.1 per million. The male-to-female ratio of 1.6 : 1 remained unchanged over the study period, as did the location and distribution of the tumors. In the United States, the incidence of osteosarcoma is considered to be higher in males than in females, occurring at a rate of 5.4 per million persons per year in males versus 4.0 per million in females, with a higher incidence in blacks (6.8 per million persons per year) and Hispanics (6.5 per million), than in whites (4.6 per million).22 The only clear change over the study period was an increase in the age of patients beyond the classic peak age range of 10 to 29 years.23 In a recent study in the United States, the incidence rates and 95% confidence intervals of osteosarcoma for all races and both sexes are 4.0 (3.5 to 4.6) for the range 0 to 14 years and 5.0 (4.6 to 5.6) for the range 0 to 19 years per year per 1 million persons. Among childhood cancers, osteosarcoma occurs eighth in general incidence. Osteosarcoma has a bimodal age distribution, having the first peak during adolescence and the second peak in older adulthood. The first peak is in the 10- to 14-year-old age group, coinciding with the pubertal growth spurt. This suggests a close relationship between the adolescent growth spurt and osteosarcoma.24 Osteosarcoma develops in 10% of patients after the age of 60 years. This group composes the second peak of the bimodal age distribution curve. In these older patients, the anatomic region of presentation differs substantially from the sites of classic osteosarcoma. Whereas lesions develop in the region of the knee (the largest and most active physes) in more than 50% of patients with classic osteosarcoma, osteosarcoma at that site develops in only 15% of the older patients. Moreover, osteosarcomas in the older population characteristically present in regions that have had previous radiotherapy, underlying Paget disease of bone, fibrous dysplasia, or some other pathological abnormality. In many ways, the older group can be thought of as having “secondary” osteosarcoma.25 An estimated 2000 malignant bone tumors are diagnosed in the United States each year. Approximately 750 of these patients have classic or conventional osteosarcomas. Males are affected slightly more often than females. Classic osteosarcoma develops in females slightly earlier than in males, and there appears to be no race predilection.26 Although the common histologic presentation of malignant cells producing osteoid would suggest a homogenous group of tumors, the morphologic appearance can vary considerably, ranging from classic osteoblastic osteosarcoma (45% of cases) through fibroblastic (9%), chondroblastic (27%), anaplastic (17%), telangiectatic, low-grade central, and other osteosarcomas (2%).27 Osteosarcoma is considered a sporadic complex genotype sarcoma, as distinguished from the balanced translocation-associated sarcomas (e.g., Ewing sarcoma). Many cell-cycle regulatory factors have been implicated, including p53, Rb, and others. Certain populations may, in fact, be at risk for developing osteosarcoma.24,28 Herein we provide a brief overview of the major regulatory pathways of the cell cycle with implications for sarcomagenesis in osteosarcoma. In conjunction with the CDK4/6 inhibitor p16INK4A, mutations of the TP53 gene are the most recurrent genetic alterations associated with cancer.29 The TP53 gene encodes the well-known tumor-suppressor p53, which plays an important role in various regulatory processes involved with cell-cycle progression. Most importantly, p53 acts as an important negative regulator, facilitating growth arrest, senescence, and/or apoptosis when cells are exposed to genotoxic, cytotoxic, and/or physiological stresses.30,31 p53 is a transcription factor possessing two transcriptional activation domains and a DNA-binding domain that recognizes specific sequences. Following exposure to cellular stress, p53 activates expression of CDKN1A, which encodes the cyclin-dependent inhibitor p21CIP1. It is through the expression of p21 that p53 negatively regulates the cell cycle.32–35 The expression level and activity of p53 is regulated by two different proteins: MDM2 and p14ARF. MDM2 is an E3 ubiquitin ligase, which facilitates the degradation of p53 in a ubiquitin- and proteasome-dependent manner.36 Interestingly, MDM2 is a transcriptional target of p53, whose expression is increased in concert with increased levels of p53.37 In addition to promoting degradation, MDM2 can also inhibit p53 function through a direct protein–protein interaction, suppressing its transcriptional activity, as well as translation of the messenger RNA transcript itself. To combat these negative regulatory effects, p14ARF is able to positively regulate p53, in part through the negative regulation of MDM2. p14ARF can actually be found at the same gene locus as CDKN2A, which encodes the CKI p16.37,38 The two gene sequences do overlap but possess alternate reading frames and are independently regulated. ARF binds MDM2, preventing it from interacting with p53.7 Consequently, p53 becomes stabilized and its overall activity increases within the cell.39 The CIP/KIP family of proteins is the second group of cyclin-dependent kinase inhibitors (CKIs). There are three different members: p21CIP1, p27KIP1, and p57KIP2. Similar to the INK4 family of CKIs, CIP/KIP proteins inhibit the kinase activity of cyclin-dependent kinases (CDKs) by preventing their association with cyclin subunits as well as adenosine triphosphate molecules, both of which are required for the phosphorylation of target substrates.32,40 Unlike the INK4 family, however, CIP/KIP proteins are able to functionally inhibit multiple CDKs. For instance, both p27 and p57 can inhibit the kinase activity of CDK4/6 and CDK2, whereas p21 acts to control the function of both CDK2 as well as CDK1.33,41 Of the three family members, p21 is the most diverse, performing a variety of functions in addition to controlling CDK activity. For example, it was previously mentioned that p21 is a transcriptional target of p53.32–35 Following exposure to various stresses, p53 activates the expression of p21, thus inducing a DNA damage response.33 In addition, p21 has been shown to localize to the cytoplasm where it acts to inhibit the induction of apoptosis by interacting with proteins involved in mediating this process. The last two cell-cycle regulators that are discussed are C-MYC and Ki-67. Both of these proteins play crucial roles during cell proliferation but do so in very different ways. C-MYC influences several processes involved in cell-cycle regulation via its function as a transcription factor. For example, C-MYC, when bound to its partner MAX, has been shown to induce the expression of cyclins D1 and D2 as well as CDK4, subsequently promoting G1 phase progression.40,42 The MYC-MAX heterodimer can also support continued cell-cycle progression through the repression of multiple CKIs, including p15, p18, p21, and p27.43 Furthermore, C-MYC can increase the expression of E2F2 and cyclin A2, both of which affect the S-phase of cell cycles and contribute to overall proliferation.45–45 Similar to C-MYC, Ki-67 has been shown to be vital for cell proliferation.46 Ki-67 is a cell proliferation-associated nuclear antigen that is thought to contribute to cell-cycle progression via its involvement in ribosomal RNA and ribosome synthesis.11,46,47 Interestingly, Ki-67 is expressed in all of the phases of cell cycle (excluding G0), but whether or not it participates in other such related processes is currently unknown. A small subset of osteosarcomas is hereditary.48 Osteosarcoma in siblings occurs in fewer than 1 in 1000 to 1 in 3000 osteosarcoma patients.49,50 Observation of two or more affected siblings in a family indicates an underlying genetic predisposition.50–57 When siblings in multiple generations are affected, an autosomal dominant disorder is most likely responsible. One example would be the hereditary form of retinoblastoma. Individuals with hereditary retinoblastoma (germline retinoblastoma gene mutation) have a 2000-fold risk for osteosarcoma in the second decade of life when compared with the general population.58–63 The gene for retinoblastoma (RB) has been localized to the long arm of chromosome 13 (13q14). The RB gene is recognized as the prototype of a tumor suppressor gene and has been implicated in the pathogenesis of a number of human neoplasms.64,65 A tumor suppressor gene normally functions by restraining cell (tumor) growth, so loss of function or inactivation of a tumor suppressor gene results in tumor growth. Loss of 13q14 (the RB gene) is thought to be responsible for the development of RB.64–68 A two-hit kinetic model for this class of genes was proposed by Knudson.69 For hereditary RB, the primary mutation in one RB locus occurs in germinal cells; for sporadically occurring RB, the primary mutation exists in somatic cells. The second step, responsible for malignant transformation, is the loss of function of the remaining normal homolog in somatic cells by some chromosomal rearrangement or mutation identified as loss of heterozygosity for markers in or around the RB gene.58,60–72 Molecular analyses of both sporadic osteosarcomas and osteosarcomas from patients with RB have revealed homozygous loss of RB gene function in a high percentage of cases.58,59,65–77 Assessment of loss of heterozygosity at the RB gene in a study by Feugeas and colleagues78 revealed that RB gene locus loss of heterozygosity could be an early predictive feature for osteosarcomas with a potentially unfavorable outcome. Osteosarcoma develops in 12% of patients with bilateral RB, yet as many as 70% of osteosarcomas have a dysfunctional RB gene product.79,80–85 Thus other oncogenes are likely implicated in the oncogenesis of osteosarcoma. Several investigators have demonstrated that the mutational profiles of the RB gene in osteosarcoma are basically the same as those for RB and that mutation of the RB gene plays an essential role in the development of osteosarcoma.65,73 Besides loss of gene function at the locus on chromosome 13, however, loss of heterozygosity for other chromosomal loci, such as 3q, 17p, and 18q, has been implicated.73,86–91 Osteosarcoma is a high-grade sarcoma comprising malignant osteoblasts that vary in size and shape and have bizarre mitoses. Proposed histologic grading systems for osteosarcoma appear to be of little value.92 Attempting to grade an osteosarcoma presents many difficulties that limit the usefulness of any grading system. For example, many tumors are heterogeneous, and tissues sampled from separate areas of the same tumor may give different impressions. The number of mitoses, the degree of cellularity, and cellular anaplasia or pleomorphism can differ from site to site within the same tumor. Tumors of identical histologic appearance often differ in their clinical behavior. All “classic” or conventional osteosarcomas are considered high-grade. Osteosarcoma is a vascular tumor, and the tumor osteoblasts produce tumor osteoid or woven bone (Fig. 92-1, A and B). These tumors are poorly differentiated and may take on a fibroblastic or chondroblastic appearance on light microscopy (Fig. 92-1, C and D), but if there are areas of bone formation, they are considered osteosarcomas. In fact, a high-grade chondroblastic sarcoma in a child or adolescent on biopsy is considered to be an osteosarcoma (and treated as such) until proved otherwise from examination of the entire specimen (chondrosarcomas are extremely unusual in children). The tumor usually originates in the metaphysis of the bone and percolates between the preexisting trabeculae of bone, incompletely destroying the existing bone, presumably because of its rapid growth (seen well in Fig. 92-1A). The tumor eventually follows the vascular Haversian and Volkmann canals in the cortex, partially resorbing the normal cortex and replacing it with tumor bone as it spreads to the adjacent soft tissue. The periosteum is lifted and tries to respond, but the response is incomplete, leading to the appearance of the Codman triangle on a radiograph. It may also cause perpendicular striations of bone, the so-called starburst appearance of osteosarcoma. Proximally, the tumor ends fairly sharply in the medullary cavity, but “skip metastases” may be detectable in the marrow surrounding the tumor in a small proportion of patients. The physis or growth plate is a relative barrier to tumor spread, but because the open physis has vascular channels, it is well documented that the tumor will cross the growth plate and enter the epiphysis. The articular cartilage is a more definitive barrier, and osteosarcomas seldom cross the articular cartilage unless there has been a fracture. It may spread into the joint at the periphery of the cartilage or enter the joint along ligaments such as the cruciate ligaments of the knee, but this is a relatively rare event (Fig. 92-2). Telangiectatic osteosarcoma is a predominantly radiolucent, destructive osteosarcoma variant. Histologically, it is composed of single or multiple dilated spaces containing blood or degenerated tumor cells and lined by anaplastic, mitotically active sarcoma cells.79 Telangiectatic osteosarcoma must be differentiated from aneurysmal bone cyst, to which it can be similar in appearance both radiographically and histologically. A review of 124 patients with telangiectatic osteosarcoma spanning the years 1921 to 1979 suggested no differences in survival compared with patients with conventional osteosarcoma. Further analysis demonstrated that the favorable outcome in 17 of the patients with telangiectatic osteosarcoma was related to their being treated with multiagent chemotherapy. Twenty-five patients had received this therapy, and 17 were free of disease at 5.5 years, demonstrating the response to chemotherapy in this highly vascular tumor.93A more recent evaluation of 323 patients with telangiectatic osteosarcoma revealed that although these tumors may be at higher risk for pathological fracture, with multimodal therapy, outcomes were similar to other osteosarcomas even with the negative prognostic factor of the fracture itself.93a The plain radiograph is the best diagnostic tool. Osteosarcomas may either completely destroy the bone (radiolucent lesion) or replace the bone with a blastic response (radiodense), but they most often do both. The radiograph shows areas of destruction of the host bone and blotchy densities of new (tumor) bone production. The lesion is most frequently in the metaphysis of a long bone in the adolescent or child and in the flat bones in the adult, but any bone can be involved at any age (Fig. 92-3, A and B). The tumor is usually large, destroys the cortex, and is associated with a soft-tissue mass. The mineralization of the matrix is often apparent, but because these tumors can contain areas of chondroblastic and fibroblastic as well as osteoblastic differentiation, the pattern on mineralization might not be that of bone. There are no blood laboratory studies that aid in the diagnosis of osteosarcoma, but it has been shown in several large series of patients that an elevated level of alkaline phosphatase and/or lactic dehydrogenase is associated with a worse prognosis.96–96 MRI is the single most useful study in evaluating the intraosseous and extraosseous extent of the primary tumor and in detecting intramedullary or transarticular “skip” metastases. It has been well demonstrated that MRI better identifies the edema (high water content) in and around the reactive zone of the pseudocapsule illuminating the potential surgical margin. MRI has also been shown to be superior to CT in displaying the medullary canal extent of the tumor, suspected “skip” lesions, soft-tissue extension, and overall anatomic location of an extremity tumor (see Fig. 92-3, D to F).97 MRI, with or without magnetic resonance angiography, is the single most valuable tool for planning limb-sparing surgical procedures.100–100 The use of MRI has significantly improved radiologic staging and presurgical planning.101,102 Serial MRI studies are less reliable, however, in evaluating tumor response to primary chemotherapy, and are more predictive of a poor rather than a good response. By demonstrating an increase in size of the tumor, more bone destruction, and soft-tissue invasion on serial studies, MRI is more accurate as a measure of a poor chemotherapy response.7,103,104 Metastases to bone and/or lung are usually assessed by a whole-body 99mTc bone scan and a complete CT of the chest and mediastinum. The 99mTc bone scan has been considered superior to other imaging studies for surveying the skeleton for metastatic or multiple lesions and for later detecting the development of skeletal metastases (see Fig. 92-3C).105 PET scans have been used in an attempt to stage and separate high-grade from low-grade tumors.106 Brenner and associates107 summarized the current usefulness of 18F-fluorodeoxyglucose (18F-FDG) PET in patients with osteosarcoma. High-resolution CT has been shown to be superior to 18F-FDG PET for detecting pulmonary metastases and is not recommended to detect bone metastases except when a suspected “skip” lesion has been identified on MRI. PET does not make it possible to differentiate between high- and low-grade osteosarcoma. It could be useful, however, in determining the appropriate area to biopsy to identify viable representative tumor tissue and in distinguishing benign aggressive lesions from other lesions where local recurrence is likely. 18F-FDG PET may be most useful to determine the response to neoadjuvant chemotherapy and to demonstrate a region of viable tumor.108 The timing between the initiation of preoperative chemotherapy and the point at which 18F-FDG PET becomes predictive remains uncertain. The response to soft-tissue postchemotherapy inflammation and healing surrounding the tumor must be characterized before this technique becomes predictive of preoperative response to chemotherapy. PET also has potential usefulness in patient follow-up, differentiating postoperative tissue changes and possible tumor recurrence especially in patients with metallic reconstructions. Orthopedic implants often hamper assessment by CT or MRI, and sequential 18F-FDG PET scans could differentiate between recurrent tumors and the normal healing process. Also, because 18F-FDG PET provides scanning of the entire patient, it may be useful when combined with CT of the chest to detect first evidence of pulmonary metastasis.109A recent study showed that PET correlates only moderately to histologic response, but may be predictive of progression-free survival.11 Once the staging workup is complete, a biopsy is performed. The procedure can be either an open biopsy or a needle biopsy (core-needle or fine-needle aspirate) depending on the experience of the surgeon, the interventional radiologist, and the pathologist. If an open biopsy is performed, it should be done by the surgeon who will be responsible for local control, with a view to placement in a site that can be resected with the definitive specimen. If a core-needle biopsy is performed by an interventional radiologist (which is becoming more frequent), the radiologist and the surgeon should agree on the placement of the needle track. Communication between the surgeon and interventional radiologist is essential, as is communication with the entire multidisciplinary team to determine whether tissue should be sent for cytogenetics or other special biological studies. Table 92-6 provides a diagnostic workup algorithm. Table 92-6 Diagnostic Workup Algorithm for Bone Sarcomas Once the biopsy results are available, the sarcoma can be staged as discussed in the introduction.
Sarcomas of Bone
Introduction
Surgical Staging System
Low (G1)
High (G2)
Parosteal osteosarcoma
Classic intramedullary osteosarcoma
Periosteal osteosarcoma (typically intermediate grade)
High-grade surface osteosarcoma
Low-grade central osteosarcoma
Paget sarcoma of bone
Radiation sarcoma
Secondary chondrosarcoma
Primary chondrosarcoma
Clear cell chondrosarcoma
Dedifferentiated chondrosarcoma
Mesenchymal chondrosarcoma
Fibrosarcoma
Fibrosarcoma
Atypical malignant fibrous histiocytoma of bone
Malignant fibrous histiocytoma (MFH) of bone
Adamantinoma
Undifferentiated primary sarcoma of bone
Giant cell sarcoma of bone
Hemangioendothelioma
Angiosarcoma
Chordoma
Neurofibrosarcoma/malignant peripheral nerve sheath tumor
Ewing sarcoma family of tumors
TX
Primary tumor cannot be assessed
T0
No evidence of primary tumor
T1
Tumor ≤8 cm in greatest dimension
T2
Tumor >8 cm in greatest dimension
T3
Discontinuous tumors in the primary bone site
GX
Grade cannot be assessed
G1
Well differentiated—low grade
G2
Moderately differentiated—low grade
G3
Poorly differentiated—high grade
G4*
Undifferentiated—high grade
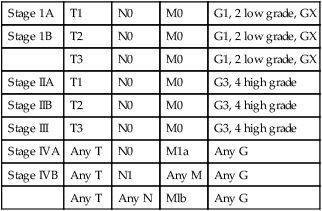
Stage 1A
T1
N0
M0
G1, 2 low grade, GX
Stage 1B
T2
N0
M0
G1, 2 low grade, GX
T3
N0
M0
G1, 2 low grade, GX
Stage IIA
T1
N0
M0
G3, 4 high grade
Stage IIB
T2
N0
M0
G3, 4 high grade
Stage III
T3
N0
M0
G3, 4 high grade
Stage IVA
Any T
N0
M1a
Any G
Stage IVB
Any T
N1
Any M
Any G
Any T
Any N
Mlb
Any G
Radiographic Staging

Staging Biopsy
Osteosarcoma
Epidemiology
Etiological and Biological Considerations
The p53-ARF-MDM2 Pathway
The CIP/KIP Family of Cyclin-Dependent Kinase Inhibitors
Other Important Players
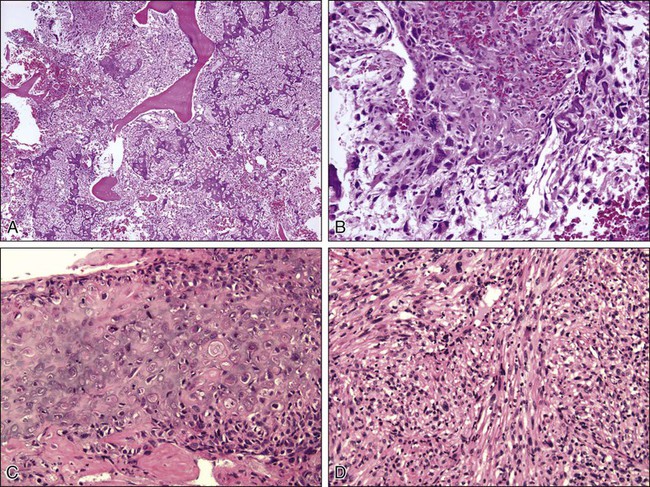
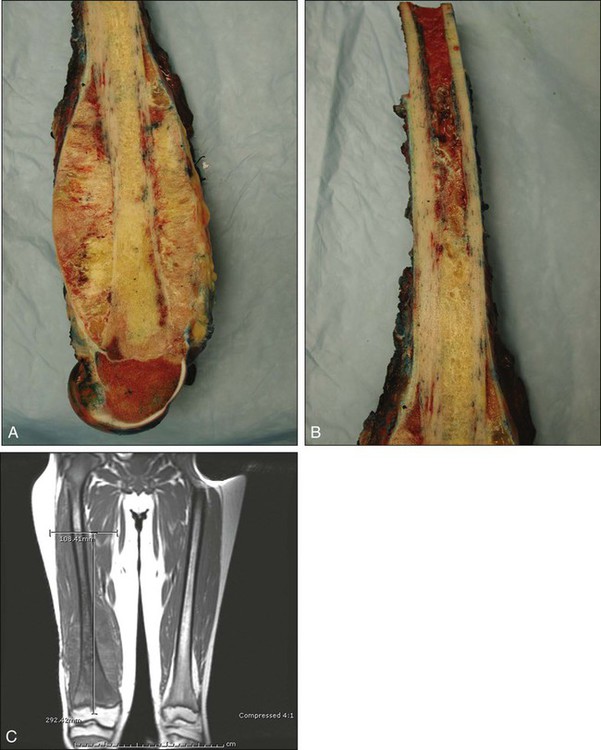
Pathology and Pathways of Spread
Clinical Manifestations, Patient Evaluation, and Staging

Sarcomas of Bone




