Fig. 3.1
Formation of BCR-AB fusion gene by reciprocal chromosomal translocation t(9;22) and their breakpoints
There are several functional domains in BCR-ABL, through which it interacts with downstream signaling molecules and transmits leukemogenic signals (Fig. 3.2). The N-terminal domain of BCR consisting from 63 amino acids with a coiled-coil motif induces tetramerization of BCR-ABL, which is essential for its biologic and leukemogenic activities [1–4]. In addition, Y177 was shown to be phosphorylated and interact with the SH2 domain of Grb2, leading to SHP-2 and Ras activation as described below. Because BCR-ABL harboring a point mutation at Y177 to phenylalanine (Y177F) can’t transform fibroblasts [5], Y177 is supposed to be one of the key sites for transforming activity of BCR-ABL through SHP-2 and/or Ras/mitogen-activated protein kinase (MAPK; also called as an extracellular signal-regulated kinase, ERK) pathways. However, this mutant BCR-ABL still could confer factor-independent growth on hematopoietic cells [6]. So, the roles of Y177 would be somewhat different between fibroblasts and hematopoietic cells probably due to the difference in the cellular context. In addition, Src-homology-2 (SH2) domain is observed in the BCR-derived region. However, this domain seems to be dispensable for its tyrosine kinase and transforming activities. BCR also has an intrinsic serine-threonine kinase domain [7], while the role of this domain in the biologic function of BCR-ABL in CML is still unknown. In addition, BCR has a Dbl homology domain with a guanine nucleotide exchange activity [8], which can activate Rho, a member of small GTPases family involved in the cytoskeleton organization.
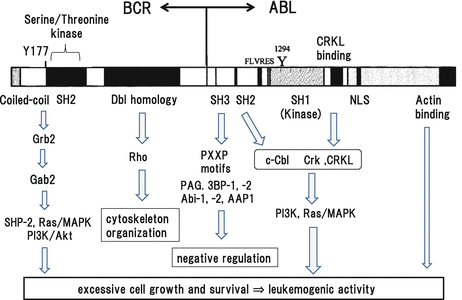
Fig. 3.2
Functional domains of BCR-ABL and its downstream molecules
The N-terminal domain of ABL closely resembles to Src family kinases in that it possesses SH3, SH2, and SH1 (kinase) domains [9]. The SH3 domain usually functions as an adaptor domain, which mediates protein-protein interactions by binding to the conserved proline-rich motif, PXXP [10]. Deletion of domain confers oncogenic activity on c-Abl [11–14]. Furthermore, a mutant BCR-ABL lacking the oligomerization domain without transforming activity recovers transforming activity by the deletion of this domain [15]. These results suggest that this domain is a negative regulatory domain for transforming activity of BCR-ABL. As for the mechanism of this negative regulation, several proteins such as PAG. 3BP-1, 3BP-2, Abi-1, Abi-2, and AAP1 that interact with this domain and inhibit c-ABL activity have been identified [10, 15–21]. However, precise role of each molecule in the biologic function of c-ABL is not clear. In addition, it remains unknown whether these molecules inhibit kinase activity of BCR-ABL as well as of c-ABL. In contrast, it was also reported that the SH3 domain can act as a positive regulator of BCR-ABL activity. A previous paper showed that, although this domain is dispensable for the transforming activity of BCR-ABL in vitro, it is required for its full oncogenic activity in vivo [22]. In this report, proliferation of the cells expressing BCR-ABL without SH3 domain was apparently reduced compared with those expressing wild-type BCR-ABL in a transplantation model, which was accompanied by the decreased binding of these cells to the extracellular matrix and infiltration into bone marrow microenvironment in the recipient mice. As for the signal transduction from this domain, this domain was shown to play an important role as a docking site for phosphorylated tyrosine residues in their partner proteins [23]. This domain is necessary for the phosphatidylinositol 3-kinase (PI3K) signaling and essential for the induction of a critical cell-cycle regulator in G1/S transition, c-myc [24, 25]. Also, this domain activates Ras/MAPK pathway [5, 6]. If this domain is deleted or mutated, the formation of BCR-ABL will be totally different. So, this mutant BCR-ABL reveals tyrosine kinase activity different from wild-type BCR-ABL through the interaction with a distinct set of cellular proteins and unable to transform fibroblasts [14, 26]. However, this mutant BCR-ABL was still capable of transforming hematopoietic cell lines [27]. Thus, functional roles of this SH2 domain would be different between fibroblasts and hematopoietic cells as was the case with Y177F as described above.
In addition to the interaction with other cellular proteins, ABL kinase activity is regulated by intraprotein (intramolecule) interactions between the SH3 domain and the linker region between the SH2 and the kinase domain as observed in Src family kinases [28–30]. Especially, the intraprotein interaction between the SH2 domain and kinase domain is both necessary and sufficient for leukemogenic activity of BCR-ABL. Disruption of this interface abrogates downstream signals and completely abolishes in vivo leukemogenic activities of BCR-ABL [31]. In addition, disruption of this SH2-kinase interface sensitizes imatinib-resistant CML cells harboring BCR-ABL mutations to TKI. Furthermore, an engineered c-Abl SH2-binding fibronectin type III monobody HA4-7c12 was shown to inhibit BCR-ABL activity in primary CML cells (Fig. 3.3), suggesting that the SH2-kinase interface can be an allosteric target to construct a new therapeutic strategy.
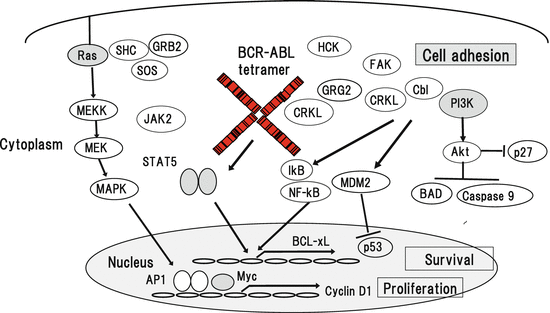
Fig. 3.3
Signal transduction from BCR-ABL to downstream pathways
The kinase domain is essential for BCR-ABL since mutants of BCR-ABL without kinase activity don’t show transforming or leukemogenic activity in any experimental models. In addition, inhibition of BCR-ABL expression by anti-sense oligonucleotides reduced proliferation of BCR-ABL-positive cell lines and suppressed colony formation from primary CML progenitors in semisolid cultures [32]. Thus, this domain plays a central role in the biologic activity of BCR-ABL and is the most appropriate therapeutic target. Based on this concept, a number of TKIs that fit in the ATP-binding pocket and competitively inhibit ATP-binding of BCR-ABL have been developed [33–35]. Among them, the 1st generation of TKI, imatinib, initially showed dramatic clinical effects in patients with CML in chronic phase compared to the conventional therapy (cytarabine + interferon-α), whereas it showed a limited degree of clinical efficacy for patients in the advanced (accelerated or blast crisis) phase [36–39]. However, about 15 % of CML patients in chronic phase still show resistance to imatinib. There are several mechanisms responsible for this resistance including amplification of the BCR-ABL gene, overexpression of BCR-ABL mRNA, point mutations of the BCR-ABL gene, and activation Src family tyrosine kinases [40–44]. Among them, point mutations of the BCR-ABL gene are most frequent and observed in 50–60 % of imatinib-resistant cases [45, 46]. At present, more than 60 point mutations of BCR-ABL have been identified, which are scattered throughout the kinase domain. To overcome these mutations, the 2nd generation of TKI (nilotinib, dasatinib, and bosutinib) have been developed and showed clinical efficacy for such cases. However, T315I mutation, which is called as a gatekeeper mutation, shows resistance to both 1st and 2nd generations of TKIs. For this mutation, the 3rd generation of TKI, ponatinib, which is effective for all types of point mutations of BCR-ABL including T315I, was developed [47, 48]. Ponatinib was approved and used in Europe and the USA, showing stable clinical efficacy for patient with CML with T315I mutation. In addition to these ATP-binding competitors, an allosteric inhibitor, GNF-2.5, which modifies the structure of the ATP-binding domain, was shown to inhibit BCR-ABL activity [49].
The kinase domain is linked to the C-terminal noncatalytic domain, which is important for biologic functions of c-Abl as well as of BCR-ABL. This domain contains the binding sites for adaptor proteins (Crk-like protein) [50, 51], a nuclear localization sequence (NLS), a DNA-binding domain [52], a p53 binding site [53], and a G- and F-actin-binding domains [1, 2, 54]. Among them, although NLS-, DNA-, and p53-binding domains are necessary for the physiological function of c-Abl, they are not required for leukemogenic activity of BCR-ABL [52, 53]. As to the role of CRKL-binding domain, a deletion mutant of BCR-ABL lacking this domain revealed reduce transformation activity by 50 % in fibroblasts [51], while it effectively conferred factor-independent growth on hematopoietic cell lines [50], suggesting the different role for this domain between hematopoetic cells and non-hematopoietic cells. The G- and F-actin-binding domain in the C-terminus is shared by another ABL family tyrosine kinase, Arg (also called as ABL2), and is indispensable for BCR-ABL to transform Rat1 fibroblasts and to confer anchorage-independent growth on NIH3T3 cells and factor-independent growth on hematopoietic cells [2, 55].
3.3 Original Function of c-ABL
c-ABL was first identified as a cellular homologue of the Abelson murine leukemia virus (v-ABL), which causes ALL/lymphoma in mice. In the v-ABL oncogene, viral gag sequences were fused to the truncated c-ABL gene lacking the NH2-terminal region that is implicated in its autoregulation. c-ABL forms a non-receptor tyrosine kinase family together with Arg, both of which have the common unique actin-binding domains in the C-terminus as describe above [56]. To examine the functional roles of c-ABL in vivo, its knockout mice were generated. However, c-ABL-deficient mice were embryonic lethal from unknown reason. Previous studies demonstrated that ABL mainly exists in the nucleus and induces apoptosis in response to DNA damage through the cooperation with the p53 homologue, p73. In addition, recent analyses showed that ABL kinases are activated by a variety of stimuli including growth factors and chemokines and the engagement of cadherin and integrin, thereby regulating cytoskeleton remodeling, cell adhesion, and migration [56]. In addition, ABL kinases are implicated in the regulation of endothelial barrier function [57, 58]. So, pharmacological inhibition of ABL kinase by imatinib reduces interstitial fluid pressure in lung and colon cancer models [8, 19, 59].
3.4 Biologic Activity of BCR-ABL
In contrast to the nuclear localization of c-ABL, BCR-ABL primarily localizes in cytoplasm. This cytoplasmic localization is necessary for its biologic activities because BCR-ABL entrapped into the nucleus by leptomycin B induces apoptosis through its tyrosine kinase activities [60]. In addition, tetramer formation is essential for BCR-ABL to reveal tyrosine kinase activity, because mutant BCR-ABL lacking the N-terminal tetramerization domain loses its transforming activities [1–4].
As for the biologic activities of BCR-ABL in leukemogenesis, a number of in vitro experiments have shown that BCR-ABL enables primitive hematopoietic cells as well as factor-dependent hematopoietic cell lines such as Ba/F3, 32D, and FDC-P1 to proliferate and survive continuously under factor-deprived conditions [61–64]. In addition, enforced expression of p210 or p190 BCR-ABL in Rat-1 fibroblasts causes a distinct morphologic change and confers both tumorigenicity and ability of anchorage-independent growth [65]. Furthermore, when bone marrow cells infected with retrovirus expressing p210 BCR-ABL are transplanted into lethally irradiated mice, recipient mice develop various types of hematologic malignancies including granulocytic hyperplasia resembling human CML, myelomonocytic leukemia, ALL, lymphomas, and erythroid leukemia [66–68]. Moreover, transgenic mice expressing p210 BCR-ABL develop pre-B or T-cell lymphomas, T-ALL, or myeloproliferative disorder like CML [69–71]. In addition, TKIs such as imatinib, nilotinib, and dasatinib are dramatically effective for CML. Together, these results indicate that BCR-ABL is a main causative gene of CML. In addition, it should be noted that BCR-ABL by itself can’t immortalize hematopoietic progenitor cells without self-renewal activity because only HSCs but not hematopoietic progenitor cells that were transfected with BCR-ABL can cause leukemia in the recipient mice in a transplantation model.
3.5 Regulation of Signaling Molecules by BCR-ABL
Growth and survival of hematopoietic cells are regulated by a number of hematopoietic growth factors. Upon the stimulation with the ligand, receptors for hematopoietic growth factors transmit mitogenic and anti-apoptotic signals through activation of intracellular downstream molecules. To keep homeostasis of hematopoiesis, these cytokine signals are subsequently eliminated by negative feedback mechanisms including ubiquitin/proteasome-dependent protein degradation, activation of phosphatases, and induction of inhibitory molecules. In contrast, activated mutants of the upstream tyrosine kinases such as tandem duplication of FLT3 (FLT3-ITD), activating point mutations of KIT, and TEL-PDGFR cause sustained activation of downstream cascades. Similarly, BCR-ABL constitutively activates various signaling molecules including Ras/ MAPK pathway, PI3K/Akt pathway, and signal transducers and activators of transcription (STATs, STAT1 and STAT5), Src family tyrosine kinases, JAK2, and so on (Fig. 3.3), thereby causing excessive growth, survival, and consequent malignant transformation of HSCs.
3.6 Ras/MAPK
Ras family proteins belong to a superfamily of GTPases that is localized to the inner surface of the plasma membrane [72, 73]. Ras family proteins play a pivotal role in a number of signaling pathways mediated by growth factors, cytokines, and cell adhesion. Activated tyrosine kinases become to associate with adapter proteins such as Shc and Grb2, which in turn recruit guanine nucleotide exchange factors (GEFs), thereby activating Ras. Once induced, Ras activates a serine/threonine kinase, Raf, which then phosphorylates MAPK kinases (also called as MEKs). Activated MAPK moves into the nucleus and phosphorylates and activates nuclear transcription factors such as Elk-1. MAPK also activates downstream kinases such as RSKs (also known as MAPK-activated protein kinases), which regulate cell-cycle regulation and apoptosis. MAPK-activated RSK catalyzes a proapoptotic protein Bad, thereby inhibiting Bad-mediated apoptosis. Furthermore, the Ras-Raf-MEK-MAPK cascade modulates cell proliferation by regulating the expression and/or activity of several proteins, including cell-cycle regulators (e.g., cyclin D1, p21WAF1, p27KIP1, and cdc25A) and transcription factors (e.g., c-fos, c-jun, and c-myc). Aberrant activation of MEK and MAPK has been demonstrated in various types of leukemic cells including CML cells [72–75].
In a previous study, we showed that induced expression of a dominant negative (DN) form of Ras (N17) inhibited the growth of p210 BCR-ABL-positive K562 cells by 90 %, while DN STAT5 (694 F) and DN PI3K (Δp85) inhibited the growth by 55 and 40 %, respectively [76]. During these cultures, the expression of cyclin D2 and cyclin D3 was suppressed by N17, 694F, and Δp85, that of cyclin E by N17, and that of cyclin A by Δp85. In addition, we found that N17 induced apoptosis in a small proportion of K562 cells, whereas 694F and Δp85 were hardly effective. During these cultures, the expression of Bcl-2 was suppressed by N17, 694F, and Δp85 and that of Bcl-XL by N17. Furthermore, although K562 was resistant to interferon-α- and dexamethasone-induced apoptosis, disruption of one pathway by N17, 694F, or Δp85 sensitized K562 to these reagents. In addition, coexpression of two DN mutants in any combinations induced severe apoptosis. These results suggest that cooperation among Ras/MAPK, STAT5, and PI3K is required for full leukemogenic activities of BCR-ABL, whereas Ras/MAPK seemed to be most important for BCR-ABL-dependent cell growth and survival.
3.7 PI3K/Akt
PI3K is another important signaling pathway controlling serine/threonine phosphorylation [74, 75, 77]. PI3K consists of two subunits, the p85 regulatory subunit and the p110 catalytic subunit. The p85 subunit binds to the ligand-activated and autophosphorylated tyrosine kinases. As a result, the p110 subunit and their downstream substrate Akt (previously called as PKB) are recruited to the membrane. PI3K/Akt pathway activates several downstream targets including p70 RSK, forkhead transcription factors (FOXOs), and NF-κB. Akt is a serine/threonine kinase and an important component of the cell survival machinery. PI3K-activated Akt provokes a number of signaling events. For example, Akt phosphorylates an NF-κB inhibitor, IκB. Upon phosphorylation, IκB is degraded by 26S proteasome and releases NF-κB, which then moves into the nucleus and induces the transcription of a number of target genes involved in cell survival such as Bcl-XL and IAPs and cell adhesion such as E-selectin, VCAM-1, and ICAM-1. Akt also phosphorylates the proapoptotic protein Bad, which leads to higher levels of free anti-apoptotic Bcl-XL, thereby inhibiting the cell-death protease, caspase-9. A tumor suppressor, PTEN, is a phosphatase that removes a phosphate from the three position of the inositol ring of the PIP3,4,5 phospholipids. PTEN has been shown to act as a negative regulator for Akt through its phosphatase activity.
The mammalian target of rapamycin (mTOR) is a serine/threonine kinase and acts as another important downstream effector of the PI3K/Akt signaling pathway, which mediates cell survival and proliferation. mTOR exists in two different complexes. mTOR forms mTORC1 by binding to Raptor, mLST8, and PRAS40, which promotes cell growth with p70 S6K as one of the main substrate. The other complex, mTORC2, is formed by the binding of mTOR to Rictor, mSIN1, and mLST8, which acts as a crucial Akt regulator.
FoxO transcription factors are human homologues of the C. elegans transcription factor DAF-16, sharing a highly conserved DNA-binding domain, forkhead box or winged-helix domain. At present, four main members (FoxO1, FoxO3a, FoxO4, and FoxO6) have been identified in the subfamily of class O. Among them, FoxO3a is considered to function as a tumor suppressor by regulating the expression of genes involved in apoptosis, cell-cycle arrest, oxidative stresses, and autophagy such as proapoptotic BIM, p27KIP1, p57, and Pdk1. Phosphorylation is the most important regulatory mechanism of FoxO3a, whereas its activity is also regulated by acetylation and ubiquitination. Although FoxO3a can be phosphorylated by several kinases, Akt is a pivotal kinase among them. When growth factors or insulin are deprived, FoxO3a translocates into the nucleus and induces the transcription of its target genes as described above, thereby inducing apoptosis and/or cell-cycle arrest (Fig. 3.4). In contrast, under the culture with growth factors or insulin, FoxO3a is phosphorylated by Akt and translocated into the cytoplasm through the interaction with 14-3-3 nuclear export protein, becoming an inactive form. Then, phosphorylated FoxO3a is trapped by an ubiquitin E3 ligase and subsequently degraded by the 26S proteasome, implying that Akt is a critical negative regulator of FoxO3a. However, a recent analysis showed that IκB kinase (IKK) also phosphorylates FoxO3a at serine 644, thereby keeping it in cytoplasm and inhibiting its transcriptional activity in an Akt-independent manner.
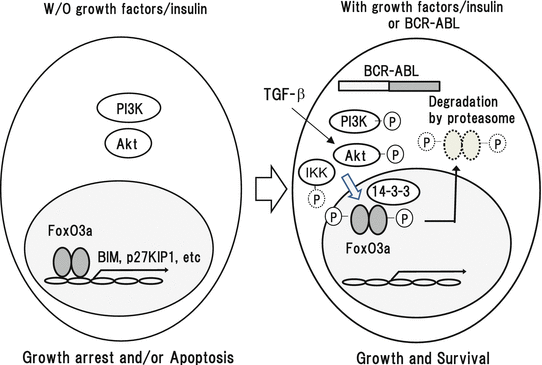
Fig. 3.4
Regulation of FoxO3a by growth factors/insulin and BCR-ABL in CML cells
With regard to the biologic roles of PI3K/Akt in CML, a mutant BCR-ABL, which can’t activate PI3K, doesn’t reveal leukemogenic activity in vitro and in vivo, indicating that PI3K/Akt pathway is essential for the leukemogenic activity of BCR-ABL [22, 78]. In addition, DN PI3K inhibits the growth and survival of BCR-ABL-positive K562 cells together with DN Ras/MAPK and DN STAT5 as described above [76]. Also, imatinib and an mTORC1 inhibitor, rapamycin, synergistically induce apoptosis in CML cells [79]. Moreover, simultaneous inhibition of mTORC1 and mTORC2 effectively induces apoptosis in CML cells with T315I mutation, for which both 1st and 2nd generations of TKIs are ineffective [80]. Furthermore, it is of clinical importance that inhibitors of PI3K/mTOR pathway sensitize CML stem cells to nilotinib, suggesting that PI3K/Akt pathway is also involved in the survival of CML stem cells [81, 82].
However, to our surprise, Akt phosphorylation was found to be reduced in CML stem cells, which resulted in the nuclear translocation (activation) of FoxO3a [83]. In this paper, serial transplantation of BCR-ABL-transformed cells originating from Foxo3a −/− mice showed decreased leukemogenic activity compared to those from Foxo3a +/+ mice, indicating that FoxO3a is essential for the maintenance of CML stem cells. Furthermore, TGF-β was shown to be a pivotal activator of Akt in CML stem cells and controls FoxO3a localization, suggesting a critical role for the TGF-β–Akt-FoxO pathway in the maintenance of CML stem cells. In contrast, it was recently reported that FoxO1 and Fox3a are inactivated and relocalized to the cytoplasm by BCR-ABL activity in CD34+ CML cells [84]. TKI treatment reduced phosphorylation of FoxOs, leading to their relocalization from cytoplasm (inactive) to nucleus (active), where they induced the expression of target genes, such as Cyclin D1, ATM, CDKN1C, and BCL6, and thereby induced G1 arrest. In addition, overexpression of FoxO3a inhibited the growth of CML cells in combination with TKI with similar results seen for inhibitors of PI3K/Akt/mTOR signaling. These data demonstrate that TKI induces G1 arrest through inhibition of the PI3K/Akt pathway and reactivation of FoxOs in CML cells. Thus, the roles for PI3KAkt/FoxO3a are rather complicated in CML cells and their stem cells. Further studies are required to clarify their precise functions, especially with attention on their crosstalk with other signaling pathways.
3.8 STATs
STATs are coded by six known mammalian genes including 10 different proteins including different isomers of STAT1, STAT3, STAT4, STAT5A, STAT5B, and STAT6 [85, 86]. Like other transcription factors, STATs have a well-defined structure including a DNA-binding domain, a conserved NH2-terminal domain, a COOH-terminal transactivation domain, and SH2 and SH3 domains. STATs mediate signals from various cytokines such as erythropoietin (EPO), thrombopoietin (TPO), granulocyte-colony stimulating factor (G-CSF), granulocyte macrophage-colony stimulating factor (GM-CSF), interleukin-2 (IL-2), IL-3, IL-5, IL-7, and IL-15. Upon tyrosine phosphorylation by upstream tyrosine kinases such as JAKs, activated STATs dimerize and translocate into the nucleus, where they activate specific target genes. A number of previous studies have shown that STATs mediate cytokine-dependent cell growth and survival by regulating the expression of cyclins, c-myc, and Bcl-XL. Among STAT family members, STAT5 is essential for the development of hematopoietic cells, because STAT5A−/−STAT5B−/− mice suffer from severe anemia and are embryonic lethal due to the disruption of EPO signaling [87]. In addition, STAT5 activity was shown to be necessary for the maintenance of HSCs [88].
STAT5 is directly activated by BCR-ABL in CML cells. Also, it can be activated by JAK2 through the mediation of AHI-1 (Fig. 3.5). Regarding the roles of STAT5 in BCR-ABL-dependent growth and survival, previous studies showed that DN STAT5 suppressed apoptosis resistance, factor-independent proliferation, and leukemogenic potential of a CML-derived cell line, K562 and BCR-ABL-transformed 32D and Ba/F3 [89–91]. In addition, a recent paper showed that STAT5B is more involved in BCR-ABL-dependent growth and survival than STAT5A, while IL-3 activates STAT5A and STAT5B equivalently [92].
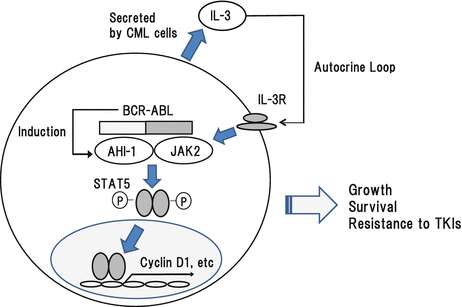
Fig. 3.5
Roles for AHI-1, JAK2, and STAT5 in CML cells
3.9 SHP-2
A tyrosine phosphatase, SHP-2, is ubiquitously expressed but highly expressed in hematopoietic cells, which has two N-terminal SH2 (N-SH2 and C-SH2) domains, a central protein tyrosine phosphatase domain, and C-terminal tyrosine phosphorylation sites. SHP-2 is involved in various signaling pathways from growth factors, cytokines, and extracellular matrix. Especially, it augments intracellular signaling from receptor tyrosine kinases. In addition, SHP-2 is known to form a stable complex with BCR-ABL, c-cbl, and Grb2-associated binder 2 (GAB2) through its SH2 domains [93, 94]. In this complex, SHP-2 is heavily tyrosine phosphorylated and plays an essential role in transforming activity of BCR-ABL for hematopoietic cells because leukemogenic activity of BCR-ABL was severely reduced in SHP-2-deficient hematopoietic cells in vitro and in vivo [95]. These results suggest that SHP-2 would be a promising therapeutic target in CML cells. To confirm this hypothesis, Sha F. et al. generated monobodies binding to the N- and C-SH2 domains of SHP-2, respectively, both of which work as competitors of SH2-phosphotyrosine interactions [96]. The monobody targeting the N-SH2 domain impaired the interaction between SHP-2 and its upstream activator, GAB2, probably by sequestering SHP-2 from the BCR-ABL protein complex. In addition, they found that inhibition of either N-SH2 or C-SH2 was sufficient to inhibit BCR-ABL-induced SHP-2 activity and to block MAPK activation, suggesting a new potent and specific therapeutic strategy for CML using the antagonists of protein-protein interactions.
3.10 Gab2
Gab2 is a member of Gab family docking proteins, which bind to receptor tyrosine kinases and other tyrosine-phosphorylated proteins via Grab2, thereby transmitting the signals to downstream molecules such as SHP-2, Ras/MAPK, PI3K/Akt, and JAK/STAT pathways. In CML cells, Grb2 binds to Y177 of BCR-ABL and bridges BCR-ABL and the C-terminal SH3 domain of Gab2. When this interaction was disrupted by Y177F mutation, BCR-ABL-dependent growth of Ba/F3 cells was severely impaired, where tyrosine phosphorylation of Gab2 and PI3K/Akt and Ras/MAPK activation were severely reduced [97]. In addition, BCR-ABL is not capable of activating PI3K/Akt and Ras/MAPK pathways effectively in Gab2-deficient hematopoietic cells [98]. Furthermore, Gab2 signaling is involved in TKI resistance in CML cells. These results indicate that Gab2 is essential for signal transductions of BCR-ABL and its leukemogenic activities [99].
3.11 c-Cbl, CRKL, and Cas (Crk-Associated Substrate)
v-Cbl was originally identified as an oncogene that causes murine leukemias. Then, later studies demonstrated that its cellular homologue, c-Cbl, regulates a number of signals from receptors for IL-2, IL-3, GM-CSF, EPO, stem cell factor, and so on through the interaction with CRKL, p85 PI3K, and Grb2 [100–103]. c-Cbl interacts with the SH2 domain of BCR-ABL [104] and phosphorylated c-Cbl binds to the SH2 domain of CRKL [105], of which complex formation is specifically observed in CML cells but not in normal hematopoietic cells. CRKL included in complex is supposed to mediate BCR-ABL signals to downstream PI3K/Akt and Ras/MAPK pathways (Fig. 3.2) [106]. In addition, a previous paper showed that inhibition of CRKL expression by antisense oligonucleotides severely reduced the growth of Ph-positive cells but not of BCR-ABL-negative hematopoietic cells [107], suggesting a key role of CRKL in the biologic activity of CML.
Cas is primarily localized at the cytoskeleton and one of the most heavily phosphorylated Crk-binding protein in v-Crk-transformed cells [108]. Cas has an SH3 domain in the N-terminus and a number of following tyrosine residues, most of which can act as potential binding sites for the SH2 domains in the partner proteins. In normal cells, Cas regulates integrin signals in combination with focal adhesion kinase (FAK) and the Src family kinases [109, 110], both of which are implicated in BCR-ABL signals. In CML cells, Cas is tyrosine phosphorylated and associates with the SH2 domain of CRKL [111]. Thus, it is supposed that Cas would transmit BCR-ABL signals to downstream molecules like c-Cbl and critically regulate the effects of BCR-ABL on cytoskeleton.
3.12 Src Family Tyrosine Kinases
Src family tyrosine kinases are first identified non-receptor tyrosine kinases including Src, Lyn, Lck, and Hck, which play crucial roles in cell growth, survival, and adhesion by mediating signals from growth factors and cytokines [112]. In addition, Src family kinases are key regulators of immune reactions in T and B lymphocytes by mediating the antigen engagement. c-Src was originally identified as a cellular homologue of the transforming oncogene Rous sarcoma virus (v-Src). v-Src activates a number of signaling proteins such as Ras/MAPK, STATs, and PI3K/Akt, thereby transforming NIH3T3 cells and conferring factor-independent growth on IL-3-dependent murine cell lines 32Dcl3 and Ba/F3 [108, 113–119]. Src family kinases are also activated in CML cells at the early stage of chronic phase in a BCR-ABL-dependent manner. However, Hck and Lyn are often overexpressed in imatinib-resistant CML cells without BCR-ABL mutations [120]. In addition, Lyn was constitutively phosphorylated and activated in imatinib-resistant CML cells without BCR-ABL mutations in a BCR-ABL-independent manner [121]. In these cells, Lyn forms a complex with the Gab2 and c-Cbl at its phosphorylation sites (Y193 and Y459), thereby inducing sustained tyrosine phosphorylation of Gab2 and BCR-ABL even in the presence of imatinib [122]. However, because no mutation was found in the kinase domain or C-terminal regulatory domain, it remains unknown how Lyn is activated in these cells [121]. In addition, suppression of Lyn expression by siRNA or pharmacological inhibition of its activity by dasatinib effectively induced cell death in imatinib-resistant CML cells, validating the use of TKIs with Src inhibitory activity such as dasatinib, bosutinib, and ponatinib for imatinib-resistant CML cases without BCR-ABL mutations.
3.13 JAK2
JAK2 is a family member of Janus family tyrosine kinases including JAK1, JAK2, and JAK3 and Tyk1 and Tyk2. This family of tyrosine kinases has tandem pseudokinase and tyrosine kinase domains in common. JAK family tyrosine kinases transmit signals from the receptor for cytokines, interferons, and hormones. Among them, JAK2 is known to mainly transmit signals from the receptors for EPO, TPO, and G-CSF [123]. In agreement with these findings, definitive hematopoiesis was severely impaired in the fetal liver of JAK2 knockout mice and these mice are embryonic lethal around day 12.5 due to severe anemia [124]. In addition, selective deletion of Jak2 in adult mouse hematopoietic cells leads to lethal anemia and thrombocytopenia [125, 126]. These results indicate that JAK2 plays a key role in the development of hematopoietic cells. Upon activation, JAK2 activates Ras/MAPK, STATs, and PI3K/Akt. In addition, JAK2 is activated in BCR-ABL-transformed cells. However, even if BCR-ABL activity is inhibited by TKI, JAK2 activity is kept in residual CML stem cells by IL-3 and G-CSF produced by themselves in the bone marrow microenvironment, thereby contributing to their resistance to TKI (Fig. 3.5) [127].
Abelson helper integration site 1 (AHI-1) is an oncogene that was identified by provirus insertional mutagenesis in v-ABL-transformed murine pre-B-cell lymphoma. Overexpression of AHI-1 augments the growth of hematopoietic cells in vitro and causes leukemia in vivo [128]. AHI-1 is highly expressed in CML stem/progenitor cells, where it forms a complex with BCR-ABL and JAK2 (Fig. 3.5) [128]. Suppression of AHI-1 expression by SiRNA reduces the growth of in CML stem/progenitor cells in vitro. In addition, it was shown that AHI-1 regulates phosphorylation status of BCR-ABL, JAK2, and STAT5 in CML cells. So, it is supposed to enhance the function of BCR-ABL. Furthermore, AHI-1-BCR-ABL-JAK2-AHI-1 complex is responsible for TKI resistance in CML stem/progenitor cells [129, 130]. These results suggest that JAK2-AHI-1 will be a potential therapeutic target in CML as well as BCR-ABL. Based on this hypothesis, Lin H et al. recently reported that dual inhibition of Jak2 and BCR-ABL by BMS-911543 in combination with TKIs effectively eliminates TKI-resistant CML stem/progenitor cells [131].
3.14 Wnt/β-Catenin
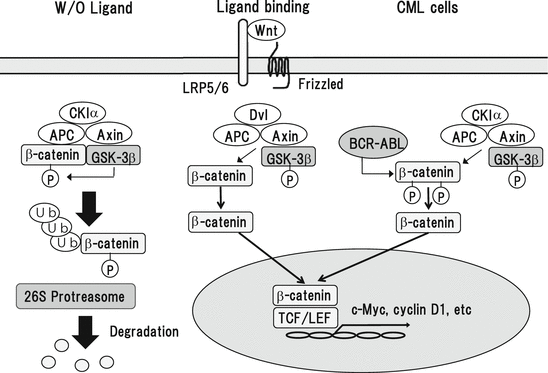
Wnt (wingless-type) ligands activate three different pathways: canonical Wnt/β-catenin, noncanonical planar cell polarity, and noncanonical Wnt/calcium pathways. The canonical Wnt/β-catenin pathway is the most studied pathway, where Wnt ligands form a complex with Frizzled (FZD) receptor and the coreceptor, low-density lipoprotein receptor-related protein 5 (LRP5) or LRP6. Without the Wnt ligand activation, β-catenin is phosphorylated by glycogen synthase kinase-3β (GSK-3β) and/or casein kinase Iα (CKIα) and subjected to degradation by ubiquitin/proteasome system (Fig. 3.6). Upon the receptor activation, phosphorylation of β-catenin is reduced and the function of “destruction complex” consisting of adenomatous polyposis coli (APC) protein and Axin is inhibited, resulting in cytoplasmic β-catenin stabilization and accumulation. Then, β-catenin translocates into the nucleus. Nuclear β-catenin acts as a transcriptional coactivator and interacts with transcriptions factors including T-cell factor (TCF) and lymphoid enhancer factor (LEF), thereby inducing the transcription of target genes such as c-Myc, cyclin D1, MMP7, and c-Jun and c-Fos family members. In addition, Wnt signaling also activates Rac, Rho, JNK, and protein kinase C (PKC) pathways through the other noncononical pathways independently of β-catenin, thereby regulating cell polarity, movement, and survival. WNT/β-catenin pathway is supposed to play an important role in self-renewal of HSCs, since enforced expression of mutant β-catenin with S33 mutation resistance to degradation is sufficient to maintain HSC population and effectively reconstitutes hematopoiesis in vivo [132]. Also, lack of WNT signals result in multilineage differentiation of HSCs [133]. These results suggest that WNT/β-catenin signaling is implicated in the maintenance of normal HSCs.
 Biomarkers for Determining the Prognosis of CML
Biomarkers for Determining the Prognosis of CML
 Molecular Mechanisms of CML Stem Cell Maintenance
Recommendations for the Management of CML in the Era of Second-Generation TKIs
Discontinuation of Therapy and Treatment-Free Remission in CML
Molecular Mechanisms of CML Stem Cell Maintenance
Recommendations for the Management of CML in the Era of Second-Generation TKIs
Discontinuation of Therapy and Treatment-Free Remission in CML
 Updated European LeukemiaNet Recommendations for the Management of CML
Updated European LeukemiaNet Recommendations for the Management of CML
 The Role of New TKIs and Combinations with Interferon-α for the Treatment of CML
The Role of New TKIs and Combinations with Interferon-α for the Treatment of CML




Related posts:
Biomarkers for Determining the Prognosis of CML
Molecular Mechanisms of CML Stem Cell Maintenance
Recommendations for the Management of CML in the Era of Second-Generation TKIs
Discontinuation of Therapy and Treatment-Free Remission in CML
Updated European LeukemiaNet Recommendations for the Management of CML
The Role of New TKIs and Combinations with Interferon-α for the Treatment of CML
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
