Histone deacetylase inhibitors (HDACI) have allowed pharmacologic manipulation of deregulated genes in cancer cells and have shown single-agent activity against T cell lymphomas, cutaneous T cell lymphomas, mantle cell lymphomas, and Hodgkin disease. The bigger promise of these agents is in enhancing the activity of other targeted therapies. In addition, the effects of HDACI on the immune system and cytokines indicate that HDACI can be useful in the treatment of immune dysfunction underlying tumorigenesis, autoimmune disorders, and graft-versus-host disease. There is also an effort to determine whether class specificity of HDACI has a biologic significance.
- •
Histone deacetylase inhibitors (HDACI) are epigenetic agents that affect the acetylation and deacetylation status of histones and other proteins, resulting in effects on gene expression and other important cellular functions.
- •
Epigenetic deregulation has been demonstrated in the pathogenesis of all types of lymphoma.
- •
HDACI as single agents have shown remarkable clinical activity in lymphomas, especially T cell lymphomas.
- •
The mechanism of the antilymphoma activity of HDACI is unknown.
- •
HDACI can be administered both orally and intravenously. They are well tolerated in the clinical setting, with a manageable side effect profile.
- •
Combining HDACI with other anticancer therapies, including cytotoxic therapies and targeted agents, is a promising new approach to the treatment of lymphomas.
Histone acetyl modifications occur in the context of nucleosomes, that are recurring packaging structures of 146 base pairs of DNA wrapped around a core of 8 histone proteins. The amino end of these histone proteins extends outwards and can be modified by chemical process like acetylation, methylation, and phosphorylation modulated by the respective set of opposing enzymes that control these chemical reactions. By affecting their secondary structure, these modifications change the spatial relationship of the histone proteins, with the DNA strand making it more or less poised for the transcription machinery to reach the DNA strand and start the process of gene transcription and protein expression. Specifically, acetylation of the ε-amino moiety on the lysine tails of histones leads to an open or transcriptionally active state of chromatin allowing transcription to proceed. In contrast, deacetylation of lysine results in a closed, condensed chromatin that prevents access of the transcription machinery to the DNA strand, thus silencing transcription. These reactions are catalyzed by 2 major classes of enzymes, referred to as HATs and HDACs. There is another class of enzymes, called histone deacetylase inhibitors (HDACIs), which can block the function of HDACs by binding to and inactivating the catalytically active pocket of HDACs. This prevents or reverses the deacetylated state of the histone and promotes transcription just like HATs. However, HDACIs are distinct from HATs and to date several compounds have been identified as having HDACI-like properties. They have therapeutic potential as anticancer agents as discussed below. There are other posttranslational modifications that can affect lysine and other amino acid residues on histones, as well as other cellular proteins, and secondarily affect their function. These modifications include methylation, ubiquitinylation, phosphorylation, glycosylation, and sumoylation. The proteins affected include, but are not limited to, transcription factors like p53, E2F, c-Myc, nuclear factor kB(NF-kB), hypoxia inducible factor (HIF-1a), estrogen, and androgen receptor complexes; DNA repair enzymes like Ku70; heat shock proteins (HSP) like HSP-90; signaling pathway intermediaries like signal transducer and activation of transcription 3 (STAT 3); and structural proteins like α-tubulin. There are several reviews on epigenetic and posttranslational modification. This article focuses on the emerging role of HDACIs in the treatment of lymphomas and multiple myeloma (MM).
Biology of HDACs
More than 18 different HDACs have been identified to date based on their homology to yeast proteins, as shown in Table1 . Class I, II, and IV HDACs require Zn 2+ as a cofactor in their active site and are generally inhibited by pan-HDACI, but data are now emerging regarding the newer HDACIs that have selective activity against specific isoenzymes (eg, tubacin is an HDACI that only blocks the action of HDAC6). Class III HDACs, also known as sirutins, are homologous to the yeast Sir 2 protein and require nicotinamide adenine dinucleotide (NAD+) as a coenzyme and are not affected by pan-HDACI. To date, there are no data to suggest that inhibiting one HDAC rather than another has any clinical benefit. The clinical significance of selective HDACs inhibition remains unclear.
| Class | Yeast Homologous Protein | HDAC Enzymes | Cellular Location | Unique Domains | Required Cofactor |
|---|---|---|---|---|---|
| I | |||||
| 1a | Rpd3 | 1,2 | Nucleus | Ubiquitously expressed | Zn 2+ |
| 1b | 3 | ||||
| 1c | 8 | ||||
| II | |||||
| IIa | Hda1 | 4,5,7 | Shuttles between nucleus and cytoplasm | — | Zn 2+ |
| IIb | 6,10 | Shuttles between nucleus and cytoplasm, contain 2 deacetylase domains, HDAC6 has α tubule deacetylase domain | — | Zn 2+ | |
| III | Sir2 | 1,2,3,4,5,6,7 | Deacetylase nonhistones and transcription factors (p53) | — | NAD+ |
| IV | 11 | — | — | Zn 2+ | |
Biology of HDACI
HDACIs are classified into 4 structural groups that vary in their potency and their ability to block various classes of HDACs, as shown in Table 2 . Besides the effects of HDAC inhibition on the acetylation status of histones, these enzymes can affect other cellular proteins, which can lead to a myriad of biologic effects downstream. Hence they should more appropriately be called protein deacetylase inhibitors. Some of the salient biologic effects of HDACI that have been observed in vitro include (1) cell cycle arrest in the G1-M, G2-M phase; (2) induction of apoptosis mediated by effects on proapoptotic and antiapoptotic mechanisms for cell death affecting both the extrinsic and intrinsic apoptotic pathways; (3) inhibition of angiogenesis; (4) increased production of reactive oxygen species (ROS) and their effects on apoptosis; (5) acetylation of tubulin and disruption of aggresome formation; (6) changes in α tubulin affecting cell motility and differentiation; (7) effects on tumor immunity via effects on T cell receptor function, cytokine milieu of immune effector cells, as well as direct upregulation of proteins on malignant cells that enhance cellular recognition by antigen presenting cells (APCs) and other immune effectors. Table 3 highlights the mechanistic pathways and proteins that are affected by HDACIs leading to the effects listed earlier. In summary, modulation of histones and other proteins alter pathways that promote proliferation, angiogenesis, differentiation, and survival in cancer cells.
| HDACI Class Potency in Vitro (IC 50 ) | Compounds | Isoenzyme Selectivity | Pharmacologic Profile |
|---|---|---|---|
| Short-chain fatty acids (mM) | Valproic acid | Class I, IIa (1, 2, 3, 8, 4, 5, 7) | Short plasma half-life, rapid metabolism, nonspecific mode of action |
| Phenylbutyrate | Class I, IIa (1, 2, 3, 8, 4, 5, 7) | ||
| Hydroxamic acids (nM) | Vorinostat (SAHA) | Class I, II HDAC (1, 2, 3, 8, 4, 5, 6, 7, 9, 10) | — |
| Belinostat (PXD 101) | Class I, II HDAC (1, 2, 3, 8, 4, 5, 6, 7, 9, 10) | ||
| LAQ824 | Class I, II HDAC (1, 2, 3, 8, 4, 5, 6, 7, 9, 10) | ||
| Panobinostat (LBH589) | Class I, II HDAC (1, 2, 3, 8, 4, 5, 6, 7, 9, 10) | ||
| Tubacin | Class IIb HDAC 6: no effect on histones, hyperacetylates α-tubulin | ||
| Benzamides (μM) | MGCD0103 | Class I HDAC (1, 2, 3, 8) | — |
| CI-994 | — | ||
| SK-7041 | — | ||
| Cyclic peptides (nM) | Romidepsin | HDAC 1, 2 > 4, 6 | — |
| MS-275 | Inhibits class >HDAC3 Does not affect HDAC 6, 8 | ||
| Sirtuin inhibitors (mM) | Niacinamide | Class II specific HDACI | — |
| Sirtinol |
| Biologic Effect | Upregulated | Downregulated | Comments |
|---|---|---|---|
| Extrinsic pathway of apoptosis (activation of caspase 8, 3, 6, 7 via external receptors on cell surface. Engages the death-induced signaling complex) | Fas, Apo/TRAIL, death receptors DR4, DR5 | cFLIP, cIAP2, and XIAP | Independent of p53 status, can overcome the antiapoptotic effect of Bcl-2 |
| Intrinsic pathway of apoptosis (activation of caspase 9, 3 from increased mitochondrial permeability and release of cytochrome C into the cytosol: tightly managed by Bcl-2 family of proteins and the BH3-only proteins) | Bax, Bak | BcL-2, BcL-xL | — |
| Cell cycle arrest: G1/M, G2/M | P27, p21, p16 | Cyclin A, cyclin D | Affects the balance between cell cycle regulators and their inhibitors like CDK4, CDK2 causing cell cycle arrest |
| Inhibits angiogenesis | Inhibitors like thrombospondin, von Hippel-Lindau factor | VEGF, hypoxia-inducible factor, surviving in vascular endothelial cells | Decreased vascularity in tumors |
| Transcription regulators | RB, CREB, p 53, BcL-6 | — | Transcriptional repression of oncogenes |
| Proliferation | JAK/STAT pathways, b-TGF pathways, p53 (proliferation), Rel A/p65 (NF-kB), Myc family of proteins affecting proliferation, HIF-1 α | — | Decreased cell growth, inhibition of oncogenes |
| Signaling mediators | Estrogen receptors, androgens, glucocorticoids | — | Decreased growth signals |
| DNA repair | KU70, FEN1, BRCA1, RAD51 | — | Increased cellular damage and activation of the apoptotic pathways |
| Chaperone proteins | Hsp-90, which affects ubiquitinylation and proteasome degradation in the acetylated state | — | Effects on proteasomes and aggresomes |
| Disruption of kinetosome assembly | Effects on the phosphorylation status of premitotic proteins | — | Affects mitosis |
| ROS | Apoptosis | — | — |
| Disruption of aggresomes | Acetylation of tubulin | — | — |
| Tumor immunity, autophagy | Effects on T cell receptor function, cytokine milieu of immune effector cells, as well as direct upregulation of proteins on malignant cells that enhance cellular recognition by APCs and other immune effectors | — | Antitumor immune response |
Biology of HDACI
HDACIs are classified into 4 structural groups that vary in their potency and their ability to block various classes of HDACs, as shown in Table 2 . Besides the effects of HDAC inhibition on the acetylation status of histones, these enzymes can affect other cellular proteins, which can lead to a myriad of biologic effects downstream. Hence they should more appropriately be called protein deacetylase inhibitors. Some of the salient biologic effects of HDACI that have been observed in vitro include (1) cell cycle arrest in the G1-M, G2-M phase; (2) induction of apoptosis mediated by effects on proapoptotic and antiapoptotic mechanisms for cell death affecting both the extrinsic and intrinsic apoptotic pathways; (3) inhibition of angiogenesis; (4) increased production of reactive oxygen species (ROS) and their effects on apoptosis; (5) acetylation of tubulin and disruption of aggresome formation; (6) changes in α tubulin affecting cell motility and differentiation; (7) effects on tumor immunity via effects on T cell receptor function, cytokine milieu of immune effector cells, as well as direct upregulation of proteins on malignant cells that enhance cellular recognition by antigen presenting cells (APCs) and other immune effectors. Table 3 highlights the mechanistic pathways and proteins that are affected by HDACIs leading to the effects listed earlier. In summary, modulation of histones and other proteins alter pathways that promote proliferation, angiogenesis, differentiation, and survival in cancer cells.
| HDACI Class Potency in Vitro (IC 50 ) | Compounds | Isoenzyme Selectivity | Pharmacologic Profile |
|---|---|---|---|
| Short-chain fatty acids (mM) | Valproic acid | Class I, IIa (1, 2, 3, 8, 4, 5, 7) | Short plasma half-life, rapid metabolism, nonspecific mode of action |
| Phenylbutyrate | Class I, IIa (1, 2, 3, 8, 4, 5, 7) | ||
| Hydroxamic acids (nM) | Vorinostat (SAHA) | Class I, II HDAC (1, 2, 3, 8, 4, 5, 6, 7, 9, 10) | — |
| Belinostat (PXD 101) | Class I, II HDAC (1, 2, 3, 8, 4, 5, 6, 7, 9, 10) | ||
| LAQ824 | Class I, II HDAC (1, 2, 3, 8, 4, 5, 6, 7, 9, 10) | ||
| Panobinostat (LBH589) | Class I, II HDAC (1, 2, 3, 8, 4, 5, 6, 7, 9, 10) | ||
| Tubacin | Class IIb HDAC 6: no effect on histones, hyperacetylates α-tubulin | ||
| Benzamides (μM) | MGCD0103 | Class I HDAC (1, 2, 3, 8) | — |
| CI-994 | — | ||
| SK-7041 | — | ||
| Cyclic peptides (nM) | Romidepsin | HDAC 1, 2 > 4, 6 | — |
| MS-275 | Inhibits class >HDAC3 Does not affect HDAC 6, 8 | ||
| Sirtuin inhibitors (mM) | Niacinamide | Class II specific HDACI | — |
| Sirtinol |
| Biologic Effect | Upregulated | Downregulated | Comments |
|---|---|---|---|
| Extrinsic pathway of apoptosis (activation of caspase 8, 3, 6, 7 via external receptors on cell surface. Engages the death-induced signaling complex) | Fas, Apo/TRAIL, death receptors DR4, DR5 | cFLIP, cIAP2, and XIAP | Independent of p53 status, can overcome the antiapoptotic effect of Bcl-2 |
| Intrinsic pathway of apoptosis (activation of caspase 9, 3 from increased mitochondrial permeability and release of cytochrome C into the cytosol: tightly managed by Bcl-2 family of proteins and the BH3-only proteins) | Bax, Bak | BcL-2, BcL-xL | — |
| Cell cycle arrest: G1/M, G2/M | P27, p21, p16 | Cyclin A, cyclin D | Affects the balance between cell cycle regulators and their inhibitors like CDK4, CDK2 causing cell cycle arrest |
| Inhibits angiogenesis | Inhibitors like thrombospondin, von Hippel-Lindau factor | VEGF, hypoxia-inducible factor, surviving in vascular endothelial cells | Decreased vascularity in tumors |
| Transcription regulators | RB, CREB, p 53, BcL-6 | — | Transcriptional repression of oncogenes |
| Proliferation | JAK/STAT pathways, b-TGF pathways, p53 (proliferation), Rel A/p65 (NF-kB), Myc family of proteins affecting proliferation, HIF-1 α | — | Decreased cell growth, inhibition of oncogenes |
| Signaling mediators | Estrogen receptors, androgens, glucocorticoids | — | Decreased growth signals |
| DNA repair | KU70, FEN1, BRCA1, RAD51 | — | Increased cellular damage and activation of the apoptotic pathways |
| Chaperone proteins | Hsp-90, which affects ubiquitinylation and proteasome degradation in the acetylated state | — | Effects on proteasomes and aggresomes |
| Disruption of kinetosome assembly | Effects on the phosphorylation status of premitotic proteins | — | Affects mitosis |
| ROS | Apoptosis | — | — |
| Disruption of aggresomes | Acetylation of tubulin | — | — |
| Tumor immunity, autophagy | Effects on T cell receptor function, cytokine milieu of immune effector cells, as well as direct upregulation of proteins on malignant cells that enhance cellular recognition by APCs and other immune effectors | — | Antitumor immune response |
Rationale for activity in lymphoid malignancies
HDACIs have shown efficacy against the treatment of lymphomas, and T cell lymphomas in particular. From a mechanistic perspective, it has been difficult to assign a mode of action of this class of drugs to any lymphoma, let alone CTCL or PTCL. Pharmacodynamic studies have demonstrated histone acetylation in peripheral blood mononuclear cells as well as tumor tissue from patients with T cell lymphoma following treatment with HDACIs; however, there is no correlation of this with clinical response to these agents. Given the pleiotropic effects of HDACI on cellular function, it is important to delineate the mechanism of their antilymphoma effects and to link them to known pathways in lymphomagenesis. The known information is discussed below.
In general, cancer cells show a high level of expression of HDAC isoenzymes and hypoacetylation of histones. This has been shown in comparisons of biopsy samples of lymphomas with normal lymphoid tissue, as well as colon cancer compared with normal colonic epithelium. Transformed cells are more sensitive to the effect of HDACI-induced apoptosis in comparison with normal cells. Aberrant expression of HDACs has been shown in lymphoid malignancies. Both cell line data and primary tissue sample studies have supported the differential expression of HDACs across reactive nodes versus lymphoma subtypes mostly involving HDAC2s. How this translates into lymphomagenesis or the sensitivity of these diseases to HDACIs is unclear. More useful data come from patient samples in various lymphoma subtypes. Class 1 HDACs (1, 2, 3, 8) were expressed in all non-Hodgkin lymphoma (NHL) and Hodgkin disease (HD) cases, whereas class II expression was variable with class 10 being present in all, but HDAC6 being present mainly in B cell lymphomas with plasmacytoid differentiation and HD. The class II HDAC6 has one of the most variable expressions among lymphoid malignancies. It has several unique properties and is known to affect the acetylation status of several proteins including α-tubulin, which is important in the regulation of microtubule stability and function. In addition, it may serve as a molecular chaperone, and plays a role in regulating the aggresome pathway that eliminates misfolded protein similar to the ubiquitin-proteasome pathway. Misfolded proteins are thought to be degraded either via the ubiquitin-proteasome pathway, or possibly through the aggresome. Misfolded proteins destined for the aggresome are thought to be transported along microtubules, via the activity of motor proteins like dynein and adapters proteins like HDAC6, to the microtubule organizing center (MTOC), which in turn transports these proteins to the lysosome for degradation as part of the HDAC6-aggresome pathway. Pharmacologic inhibition of HDAC6 results in hyperacetylation of tubulin and disruption of the aggresome-mediated pathway resulting in apoptosis, which may also explain the mechanistic basis for their synergy with proteasome inhibitors (PI) as discussed below. Overexpression of HDACs may result in the decreased expression of tumor suppressor genes, leading to carcinogenesis. One oncogenic mechanism that may involve aberrant HDAC expression includes the recruitment of HDACs to promoter regions of key genetic sequences. It has been shown that specific chromosomal translocations in leukemias lead to expression of oncogenic fusion proteins that form aberrant association with HDACs at promoter site genes and leading to the onset of tumorigenesis. This process is exemplified by the translocation t (15,17) in acute promyelocytic leukemia (APL) that results in the fusion promyelocytic leukemia–retinoic acid receptor (PML-RAR) (APL), that recruits the HDAC3-containing repressor complexes leading to a decreased expression for differentiation-specific genes. Similarly chromosomal translocations resulting in the recruitment of class I HDACs to oncogenic promoter sites may underlie the pathogenesis of T cell NHL. Overexpression of repressive transcription factors that interact with HDACs and affect the promoter regions of tumor suppressor genes may also underlie the pathogenesis of hematological malignancies.
Tumor microenvironment and tumor immunology play important parts in the pathogenesis and progression of most lymphomas and HD. HDACs and HDACIs are shown to be important regulators of the immune response and induction of tolerance. Villagra and colleagues have demonstrated that overexpression of HDAC11 inhibits interleukin (IL) 10 expression and induces inflammatory antigen presenting cells that are able to prime naïve T cells and restore the responsiveness of tolerant CD4+ cells. Disruption of HDAC11 in APCs leads to the upregulation of expression of the gene encoding for IL-10, leading to impairment of T cell responses, which may contribute to their antilymphoma effects.
There is likely to be further delineation of the effects of various HDACIs on specific cellular functions. There is currently no evidence that inhibiting one HDAC enzyme rather than another is associated with improved activity, or that more selective HDACIs will be associated with an improved adverse effects profile.
Further discussion of HDACIs in specific lymphoma subtypes is presented below.
HDAC Modulation of T Cell Lymphomas
CTCL is the disease with the highest clinical response rates with HDACIs. A large study of the expression of various HDACs in 73 patient samples has correlated this with the clinical behavior. The expression of HDAC1 and HDAC6 was similar between indolent and aggressive cases but the expression of HDAC2 and acetylated histone 4 (H4) was higher in cases of aggressive disease (HDAC2 55.5% aggressive CTCL vs 15% indolent, acetylated H4 22% aggressive vs 8%). Survival correlated with the overall expression of HDAC6 (hazard ratio 0.39) independently of the CTCL subtype. Other epigenetic markers were also deregulated in CTCL. Van Doorn and colleagues compared genome wide DNA methylation screening in samples of CTCL with benign skin disorders. They showed widespread promoter hypermethylation in malignant T cells in CTCL, suggesting epigenetic instability. Specific CpG islands of more than 35 promoter regions were hypermethylated including the tumor suppressor gene BCL7a (B cell chronic lymphocytic leukemia [CLL]/lymphoma) in 48% of samples, PTPRG (protein tyrosine phosphatase receptor γ) gene in 27% of samples and THBS4 gene thrombospondin 4) in 52% of patient samples. These genes were also hypermethylated in the CTCL cell lines but not in the control samples. BCL7, located on chromosome 12q 24.3, is of particular importance because it has been cloned as part of the chromosomal translocations seen in Burkitt lymphoma. Its expression is diminished in mycosis fungoides and PTCL compared with lymphoblastic lymphoma. Other genes that were hypermethylated in CTCL samples compared with normal skin are grouped into the following categories: cell cycle deregulation (p15, p16, p73), defective DNA repair genes (MGMT), apoptosis deregulation (TMSI, p73), and chromosomal instability (CHFR). Hypermethylation of p73 has also been described in nodal B cell lymphomas and natural killer (NK) cell lymphomas. Promoter hypermethylation of P16 has also been noted in CD30+ T cell NHL.
For further mechanistic insight into the action of HDACIs in CTCL, Duvic and colleagues attempted to look at biologic correlatives of HDACI therapy in patients with CTCL by performing serial skin biopsies on patients receiving vorinostat on trial at 2 hours, 4 hours, 8 hours, and then 12 weeks after initiation of treatment. These results established the following: at 4 weeks, 39% of the patient samples showed lymphocyte depletion consistent with the antilymphoma effect of vorinostat. At 4 weeks after therapy, there was a decrease in dermal microvessel density as measured by CD31 positivity on dermal vessels in all patients, but this was significantly lower in responding patients ( P = .001). Prior cell line data using the CTCL cell line HH indicated that a 24-hour exposure to vorinostat resulted in an 8-fold increase of the antiangiogenic protein TSP-1 as studied by gene expression array. Consistent with the cell line data, an increase in the dermal TSP-1 staining was noted as early as 2 hours after treatment and was present at 8 weeks in 6 of the 17 paired lesions, including 4 of the 6 responders. Another important protein that is constitutively activated in CTCL is phsophorylated STAT-3 (p-STAT3), which can be detected by immunohistochemical stains either in the nucleus or cytoplasm within both the keratinocytes and the lymphocytes in the lesions. In this study, nuclear staining for p-STAT3 was prominent in both keratinocytes and lymphocytes before the start of therapy. After 4 weeks of therapy with vorinostat, the staining pattern shifted to localization within the cytoplasm (inactive state) in 9 of the 11 patients who responded, whereas this shift was noted in only 3 of the 16 nonresponders. This shift was noted as early as 2 hours after treatment in 4 of 11 paired lesions. Using a similar model of paired skin biopsies, Ellis and colleagues performed gene expression profiles (GEP) and real-time quantitative polymerase chain reaction (PCR) on skin samples from 6 patients with CTCL who were being treated with panobinostat in a phase I trial. These biopsies were obtained at 0, 4, 8, and 24 hours after administration of drug. In this study, there were 10 patients with a diagnosis of relapsed CTCL who were treated at varying dose levels as part of a large phase I study in patients with hematological malignancies. Clinical efficacy was observed in 8 patients (2 achieved a complete remission-CR at both dose levels and 4 patients achieved a pa partial remission-PR, 2 patients had stable disease-SD). The skin biopsy data showed that there was hyperacetylation of histone H3 in tumor cells as early as 4 hours after treatment. Consistent with previous data, histone acetylation within mononuclear cells was shown in both responders and nonresponders up to 48 to 72 hours after the last oral dose, indicating that this could not be used as a therapeutic marker. GEP data from all 6 patients consistently showed that panobinostat induced transcriptional regression of a greater number of genes than activation. The genes that were consistently affected included genes affecting cell cycle (CCNDI, IGFI) apoptosis (septin10, TEF, SORBBS2), angiogenesis (GUCY1A1, ANGPT1), and immune modulation (LAIR1). CDKN1A, which codes for p21, was unregulated in response to HDACI therapy, although in this study upregulation of p21 was not consistently seen in all patients. Of the 23 genes, 4 were further selected for validation by QRT-PCR. These data confirmed downregulation of guanylate cyclase 1A3 (GUCY1A3), the proangiogenic gene ANGPT1a, and the transcription factor COUP-TFII (NR2F20, which is an upstream regulator of ANGPT1 and CCND1). These effects on genes controlling angiogenesis are consistent with the effects noted by Duvic and colleagues and provide confirmation that the antiangiogenic effects of HDACI therapy may be important in their mechanism of action. Bates and colleagues conducted a clinical trial of romidepsin in patients with PTCL and CTCL and also studied the biologic correlates of activity of HDACI in peripheral blood and tumor samples. Predetermined markers of HDACI activity included global histone acetylation and expression of the ABCB1 gene (encodes for the p-glycoprotein called mixed drug resistance) and fetal hemoglobin. The histone acetylation data correlated with pharmakokinetics parameters of area under the curve (AUC) and maximum plasma concentration C max , though there was no correlation between response, histone acetylation, and the expression of either ABCB1 or fetal hemoglobin, indicating the need for improved biomarkers to predict responses with HDACI.
HDACIs an affect signaling patterns from cell surface receptors and, increasingly, T cell malignancies are associated with deregulation of the T cell receptor (TCR) signaling and the immune function. Investigations into the effects of HDACIs, particularly vorinostat have been conducted on TCR signaling and the immune system to delineate more specific mechanisms of action for this agent, as well as to understand the basis for combining it with other agents. Wozniak and colleagues performed extensive studies using GEP on a panel of CTCL cell lines (HH, HUT78, MJ, Myla, SeAx) that were exposed to vorinostat at various time points. The functional analysis of these altered genes revealed pathways including cycle regulators for G1/S transition (E2F, E2F4, cyclin-dependent kinase [CDK] 4, CDK6, cyclin A2, D2, D3, E20), G2/M regulators (CDC23, CD25B, and CHEK4), apoptosis (FAS, IRAK1, CASP6, BID, BCL2), antiproliferative genes, as well as multiple mitogen-activated signaling kinase (MAPK) signaling pathway (MAPK1, MAP3K6, MAP3K14) as described earlier. However, this study also showed changes in genes that are involved in the JAK/STAT signaling pathway, cytokine-cytokine interaction, and expression of receptors belonging to the tumor necrosis factor (TNF) family, all important pathways for survival and differentiation of lymphocytes and the immune system. Vorinostat treatment was shown to shift the expression profile of cytokines, resulting in increased expression of IL-1a, IL-6, and Il-9, and a decrease in the expression of IL-4, IL-5, Il-10, IL-11, and their associated receptors. Overall, the cytokine profile represented a state that inhibited lymphocyte growth and proliferation and inhibited the TH2-type immune responses. The latter aspect of the drug effect is important because CTCL is a malignancy of activated T cells and is characterized by deregulation of the immune system with reversal of the Th1/Th2 cytokine profile. Vorinostat has also been shown to affect genes that affect cell migration and chemotaxis, which may affect the skin homing properties of malignant cells in CTCL including a decrease in the expression of cytokine genes like CCL1, CCL22, CXCL10, CCR4, and CCR6 and an increase in others like CCR2 and CCR6. There was also some alteration in the expression of members of the JAK/STAT pathway like STAT6, STAT5A, and SOCS2 (decrease), and STAT1, STAT3, and JAK1 (increase).
The TCR signaling pathway is crucial for the survival of T cells and is altered in T cell malignancies. HDACI have been shown to modify this pathway. In general, antigen stimulation engages TCR signaling and induces the recruitment of several kinases including lymphocyte-specific protein tyrosine kinase and a protooncogne named FYN, resulting in phosphorylation of many downstream substrates including CD3 chains, TCR ε chain, and the ζ chain associated with ZAP-70 and phospholipase C. Several downstream pathways that are activated include the PKC, MAPK/p38, Jun pathway and the serine-threonine protein kinase PI3K/AKT pathway, which is important for the survival of T lymphocytes. Treatment with vorinostat induces repression of all genes associated with TCR-related signaling including ZAP-70, CD3DIL4, IL-5, Il-10, and FOXP3, and upregulates FYN, interferon γ, and IL-12A. The effects on TCR signaling were significant and were seen across all cell lines and confirmed by QT-PCR. A decrease in the phosphorylated forms of ZAP-70 and AKT after vorinostat treatment was confirmed by Western blots, confirming the inhibitory effects of this agent on TCR signaling. FYN, which is upregulated by vorinostat, is an tyrosine kinase that belongs to the SRC family kinases that phosphorylates several negative regulators of TCR signaling and ultimately adds to the negative effect of vorinostat on TCR signaling pathways.
In summary, there are many signaling pathways that may be altered in T cell NHL, and HDACI seem to affect them in a myriad of different ways. As this knowledge of this increases, it will be logical to combine multiple targeted agents to optimize the antilymphoma effects of these agents.
HDAC Modulation of B Cell Lymphomas
Presently, there are no HDACIs that are approved specifically for the treatment of B cell lymphomas, although there is a strong rationale for their use in B cell malignancies. Mantle cell lymphoma (MCL) is considered to be incurable with known therapies and is a unique disease characterized by marked deregulation of cyclin D1 mediated by the t(11:14) translocation and loss of the CDK inhibitors p21 and p27. Two of the most prominent effects of HDACI are the downregulation of cyclin D1 and upregulation of p21/p27. The clinical data of HDACI in MCL are promising and represent an interesting avenue for combination therapy to treat this disease. Similarly, the association of deregulated BCL-6 in many cases of diffuse large B cell lymphoma (DLBCL) and the effects of HDACI on BCL-6 provide a rationale for the use of HDACI either alone or in combination with other agents in cases of lymphoma in which BCL-6 is over expressed. DLBCL is one of the most common subtypes of NHL and has 2 subtypes based on gene expression analysis. The more common subtype is the germinal center (GC) subtype that overexpresses BCL-6 and CD10, whereas the activated B cell (ABC) subtype expresses activation markers like MUM1 and/or CD138. The ABC subtype has low levels of BCL-6 but high levels of NF-kB and STAT3 and is more chemorefractory, leading to a worse prognosis for the patients. The overexpression of the transcription factor Bcl-6 in DLBCL (GC) from chromosomal translocations leads to recruitment of several HDACs including HDAC1, 2, 4, 5, and 7, which causes the repression of growth-regulatory target genes like p53, p21, and STAT3. Bcl6 can be inhibited by acetylation (through HDACI therapy as well as through inhibition of SIR-2) and leads to the activation and expression of p53, resulting in downstream effects like apoptosis. In ABC subtypes, HDACI can result in decreased expression of STAT3 through its association with HDAC1, resulting in inhibition of activated STAT3 and its dephosphorylation, leading to growth inhibition of this subtype as well. STAT3 is also a transcriptional target of Bcl-6 but, in contrast with p53, it functions as an oncogene. These data provide a rationale for the use of HDACI in DLBCL (both GC and ABC subtypes) and has formed the basis of an ongoing trial using a combination of HDACI and Sir-2 inhibitors for the treatment of GC B cell lymphomas.
HDAC Modulation of Hodgkin’s Disease
In vitro data support the potential activity of HDACIs against HD cell lines. Biologically, HD is characterized by a high level of cytokine secretion from the inflammatory infiltrate surrounding the pathognomonic Reed-Sternberg (RS) cells. Therapeutic strategies include targeting the malignant cells as well as the inflammatory milieu and cytokines including, IL-5, IL-6, IL-7, IL-9, IL-10, IL-13, and thymus activation–regulated chemokine (TARC/CCL17), which are important in the pathogenesis of HD. Many of these are part of an autocrine loop thought to activate the JAK/STAT pathways, resulting in continual activation of the STAT family of transcription proteins, in particular STAT3 and STAT6. Cytokines including IL-2, IL-6, IL-7, IL-9, IL-10, and IL-15 induce the activation of STAT3, whereas STAT6 is primarily induced by IL4 and IL-13 and may depend on an autocrine IL-13 loop secreted by RS cells. Phosphorylated STAT6 localizes to the nucleus and induces the expression of STAT6 target genes that include TARC and IL-13 as well as other cytokines that attract TH2-specific lymphocytes into the tumor microenvironment. These lymphocytes are involved in humoral immunity and promote allergic responses, which suggests the importance of STAT6 in the survival of RS cells as well as promoting the unique cellular and immunologic milieu that is a hallmark of HD. STAT regulation involves phosphorylation as well as lysine acetylation, which implies that HDACIs could play a role in regulating critical features of HD biology. This is supported by data from Buglio and colleagues, where they demonstrated that exposure of HD cells (L-428 and KM-H2) to vorinostat resulted in an increase in histone acetylation and p21 expression, and caspase-mediated apoptosis. Vorinostat selectively inhibited STAT6 phosphorylation and resulted in decreased mRNA levels of STAT6 seen by PCR and a reduction of TARC as a downstream effect. Changes in cytokines were evaluated in the supernatants of exposed cells showing an increase in the level of IL-13 and interferon inducible protein (IP)-10, and a significant decrease in the level of IL-5, confirming a shift in the Th1/Th2 cytokine balance. Another target gene regulated by STAT6 is the antiapoptotic Bcl-xL, which was significantly decreased in the HD cell lines following vorinostat exposure. This reduced level of Bcl-xL could reduce the apoptotic threshold enough to allow synergistic activity with other anti-HD agents including chemotherapy. Combinations of HDACI with hypomethylating agent can also influence antitumor immune responses by affecting the expression of proteins like the cancer testis antigens (CTA), which include MAGE, SSX, and NY-ESO, in a variety of tumors including Hodgkin lymphoma (HL). These immunomodulatory effects may underlie the therapeutic activity of these agents in HL.
HDAC Modulation of MM
In contrast with lymphomas, there are no studies showing abnormal expression of HDACs in plasma cell malignancies like myeloma. In spite of this, several HDACIs have shown antimyeloma activity. The mechanism of this activity remains unclear but may be related to the general effects of HDACIs on cell biology, like upregulation of p21, cell cycle arrest, or apoptosis. Preclinical data support antimyeloma activity with HDACI, as shown by the modulation of gene expression in MM cells by HDACIs. Both suberoylanilide hydroxamic acid (SAHA) and valproic acid (VPA) alter the expression of oncogenes, cell cycle regulators, antiapoptotic transcription factors, and members of the IGF-IR and IL-6R signaling cascades, important in the pathogenesis and progression of MM. In addition, VPA has been shown to alter genes that contribute to RNA splicing and transcription as well as DNA replication, indicating effects on cell growth that are independent of cell cycle regulation and apoptotic pathways. These data are consistent with the effects of HDACIs shown in other cell lines.
Direct cytotoxic effects of HDACIs have been reported in various myeloma cell lines, indicating differences in potency between the different agents: sodium butyrate and valproate acid being the least potent (inhibitory concentration of 50% [IC 50 ] in mM), FK228 and LBH589 are the most potent in myeloma. The antimyeloma apoptotic effect is independent of IL-6, a key growth factor for MM cells. Coculturing the MM cells with bone marrow stromal cells did not protect the cells from death, indicating that HDACIs could overcome the protective effect of the stromal microenvironment, which is one of the key clinical issues in MM. Mechanistically, the antimyeloma effects of HDACIs seems to involve both the extrinsic and intrinsic apoptotic pathways. MM cells contain high levels of Bcl-2 and Mcl-1 and lower levels of Bax compared with normal plasma cells, making them more resistant to apoptosis. VPA resulted in the redistribution of death receptor (DR4) to lipid rafts, resulting in improved DR4-related signaling and restoring the sensitivity of U266 MM cell lines to APO21/TNF-related apoptosis-inducing ligand (TRAIL)–induced apoptosis. LBH586 resulted in the activation of caspase 8 and the downregulation of the gene TOSA, which is a negative regulator of Fas ligand (FasL). Treatment with LBH589 caused apoptosis of the cell line MMIS (malignant melanoma in situ) by affecting the translocation of mitochondrial proteins like cytochrome c and apoptosis-inducing factor; upregulation of Apaf-1; and cleaving of Bid, caspase 9, and caspase 3. SAHA also decreased the expression of the antiapoptotic protein FLICE-like inhibitory protein (FLIP) and other members of the inhibitors of apoptosis (IAP) family such as X-linked IAP (XIAP). This process resulted in the sensitization of the MM cells (MMIS) to a Fas-activating monoclonal antibody (CH-11) and to recombinant TRAIL. Treatment of the MM cell lines U266, as well as primary myeloma cell lines, with HDACIs resulted in decreased expression of the antiapoptotic proteins Mcl-1, Bcl2, and Bcl-xL, and an increase in Bax. Both LBH589 and SAHA resulted in poly-ADP-ribose (PARP) cleavage in MM cells by 2 distinct mechanisms involving caspase 3 and calpain. Overexpression of the antiapoptotic protein Bcl-2 inhibited SAHA-induced apoptosis in MM cells. Another HDACI named KD5170 resulted in Bax activation and cleavage of caspases 9 and 3, resulting in activation of the intrinsic apoptotic system in U266 cell lines. Autophagy is another method of cell death and HDACIs have been shown to affect this pathway as well. Schwartz and colleagues showed that VPA-treated myeloma cells had cleavage of caspase 3 and autophagy granules, the first observation of this nature.
All HDACIs except tubacin induce cell cycle arrest in G1/S phase by affecting cyclins, especially cyclin D, and their CDKs. The balance is maintained by the balance between these kinases and their inhibitors like p16, p21, and p27. In MM, constitutive phosphorylation of the Rb protein may be fundamental to the growth and development of the tumor, as indicated by the increased levels of cyclins in G1compared with healthy plasma cells. In MM, HDACI-associated cell cycle arrest is associated with induction of p21 as well as a reduction of cyclin D1 and D2, thus affecting the transition of cells from G1 to S and resulting in arrest at this stage.
Another important antimyeloma action of HDACI involves degradation of misfolded proteins in the cell via the ubiquitination-proteasome and the aggresomal protein. The aggresomal system is particularly important in MM. Aggresomes are formed by the retrograde transport of misfolded proteins on microtubules and travel to the MTOC where they are sequestered for lysosomal degradation. Movement along the microtubules involves intact microtubules and the motor dynein. HDACs deacetylate α-tubulin and play a key role in the aggresomal pathway by affecting the motility of proteins along the microtubule. Targeting HDAC6 with tubacin or a pan-HDACI such as vorinostat or LBH586 results in hyperacetylation of the α-tubulin, accumulation of polyubiquitinated proteins, and apoptosis. Tubucin can inhibit the growth of MM cell lines, both drug-sensitive (eg, MMIS, U266, INA-6, and RPMI8226) and drug-resistant cell lines (eg, RPMI-LR5 and RPMI-Dox40) with an IC 50 between 5 and 20 μM, but no cytotoxicity is seen in normal peripheral mononuclear cells. Thus tubacin selectively targets malignant cells independently of the drug resistance state of these cells. The linkage between inhibition of an HDAC and the proteasome pathway has raised a strong mechanistic rational for the combination of these agents in the clinical setting, much of which is centered on the unfolded or misfolded protein response.
Vorinostat has been shown to suppress the expression of receptor genes involved in MM cell proliferation, survival, and migration, like IGF-1R, IL-6R and its key signal transducer gp130, TNF-R, CD138 (syndecan-1), and CXCR-4. Vorinostat also suppressed the autocrine IGF-1 production and paracrine IL-6 secretion of bone marrow stem cells by triggering MM (MMIS) cell binding, suggesting that it can overcome cell adhesion–mediated resistance. NaB reduced IL-6R in cell lines. Increased p21 expression and apoptosis were observed in these cell lines along with lines that were transfected with an expression vector of IL6-R, indicating that downregulation of IL-6R is not required for HDACI-induced apoptosis, again emphasizing the many pathways affected by HDACIs in MM cells.
HDACIs also affects angiogenesis in MM by a direct effect on growth and differentiation of endothelial cells and the downregulation of proangiogenic genes in tumor cells. Using OPM-2 and KM3 cell lines, VPA has been shown to decrease vascular endothelial growth factor (VEGF) secretion and VEGF receptor expression, resulting in inhibition of the vascular tubule formation in endothelial cells in cocultures with myeloma cells.
De Bruyne and colleagues showed that CD9, a tetraspanin, shows an inverse correlation between its expression level and tumor metastasis in solid tumors. In MM, CD9 is downregulated and treatment with LBH589 could result in its upregulation, making it more susceptible to NK cell–mediated cytolysis. Its expression is correlated with nonactive MM disease. This finding indicates that the immunologically mediated effects of HDACI are also importation in MM.
Pharmacology of HDACI
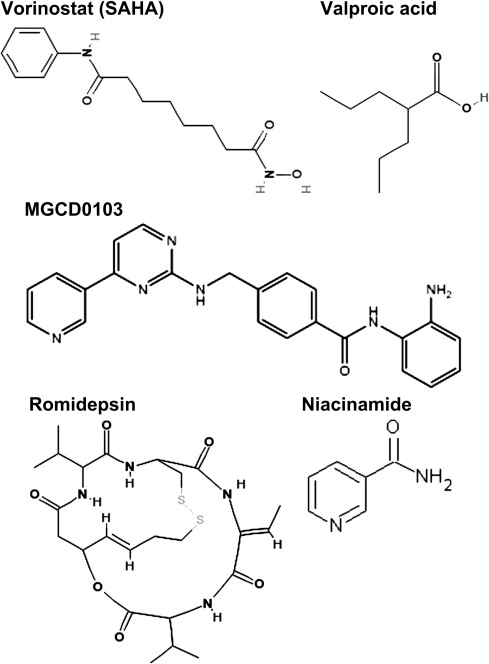
HDACIs are a chemically diverse group of naturally occurring and synthetic molecules ( Fig. 1 ) that inhibit the activity of HDACs in a wide range of concentrations from low nanomolar to high millimolar, as listed in Table 3 . Presently, there are at least 18 different HDACI being evaluated in preclinical and early clinical trials that target the Zn 2+ -dependent (ie, class 1, 2, and 4) HDACs. Most HDACI have a short plasma half-life (vorinostat, 2 hours ; romidepsin, 3 hours ; MGCD0103, 9 hours ) and undergo hepatic metabolism either via the CYP450 system (romidepsin) or the glucuronidation system (vorinostat). The metabolites are excreted through the biliary and fecal routes. HDACIs have varying formulations and differing side effect profiles depending on the route of administration. The 2 agents approved by the US Food and Drug Administration (FDA) to date (namely vorinostat and romidepsin) have undergone extensive pharmacokinetic monitoring as part of the clinical trials that led to their approval. One of the earliest pharmacologic studies was performed with vorinostat, first with the intravenous (IV) preparation and then with the oral formulation. A direct comparison of the toxicity and pharmacokinetic profiles of IV versus oral vorinostat revealed that the C max of exposure to vorinostat was higher with the IV formulation versus oral formulation (2408 ng/mL vs 658 ng/mL) but the AUC was greater with the oral route of exposure (4634 h × ng/mL vs 101,854 h × ng/mL). The toxicity profiles of these 2 regimens were also different, with more thrombocytopenia, dehydration, and diarrhea noted with the oral formulation compared with the IV. Oral vorinostat (the approved formulation) is 71% bound to proteins and is metabolized via glucuronidation and hydrolysis followed by β-oxidation into 2 inactive metabolites. Biotransformation by cytochrome P450 is negligible. It is excreted by the kidneys with less than 1% of the dose recovered as unchanged drug in the urine. The mean urinary excretion of the 2 pharmacologically inactive metabolites at steady state was 52% (±13.3%) of the vorinostat dose; 16% (±5.8%) of the dose as O-glucuronide and 36% (±8.6%) of the dose as 4-anilino-4-oxobutanoic acid. Romidepsin is highly protein bound in plasma (92%–94%) over the concentration range of 50 ng/mL to 1000 ng/mL with α1-acid-glycoprotein (AAG) being the principal binding protein. In contrast with vorinostat, it undergoes extensive metabolism in vitro, primarily by CYP3A4 with minor contributions from CYP3A5, CYP1A1, CYP2B6, and CYP2C19. At therapeutic concentrations, romidepsin did not competitively inhibit CYP1A2, CYP2C9, CYP2C19, CYP2D6, CYP2E1, or CYP3A4 in vitro.