Species
V segment usage
Common VγVδ usage
V(d) J diversity
Day of exportation from the embryonic thymus
Distribution
Mouse
Vγ1
Vγ1Vδ6.3 (liver)
High
From E18 onward
Spleen, liver
Vγ4
High
From E15 onward
Spleen, liver, lung
Vγ5
Vγ5Vδ1
Invariant
From E15 until E17
Epidermis
Vγ6
Vγ6Vδ1 (uterovaginal epithelia)
Invariant
From E16 until E18
Liver, lung, uterovaginal epithelia, tongue
Vγ7
Vγ7Vδ4
Intermediate
Not applicable (extra-thymic development)
Gut epithelia
Vγ7Vδ5
Vγ7Vδ6
Human
Vδ1
High
Unknown
Spleen, liver, epithelia, dermis
Vδ2
Vγ9Vδ2
Intermediate
Unknown
Peripheral blood
Vδ3
High
Unknown
Liver, gut epithelia
Even though a great diversity of TCRγδ can be theoretically generated in rodents and humans, the set of TCRs detected on peripheral γδ T cells is far more limited. Individual γδ T-cell subsets in particular tissue locations show biased use of certain TCR V gene segments and, in some cases, express “invariant” TCR with identical (canonical) junctional sequences [5] (Table 13.1).
13.2.1 Mouse γδ T-Cell Subsets
Murine γδ T cells are generated in the thymus in “developmental waves” that sequentially populate different tissues by regulated expression of appropriate chemokine receptors (Table 13.1). Mouse thymocytes bearing an invariant canonical Vγ5Vδ1 TCR at embryonic day E15–17 are the first to leave the fetal thymus, giving rise to skin-associated dendritic epidermal T cells (DETCs); thymocytes bearing a Vγ6Jγ1Cγ1 TCR at E16–18 give rise to the γδ T cells in the tongue and reproductive tract; peri- and postnatal thymocytes bearing Vγ1Cγ1 and Vγ4Cγ1 TCRs give rise to systemic γδ T cells. This sequential generation of γδ T cells at different stages of ontogeny is a fixed developmental program; for example, the disruption of the generation of γδ T cells in the early fetal thymus by the administration of an anti-γδ-TCR antibody to pregnant mice resulted in selective absence of DETCs in adult mice [7].
It is thought that the highly restricted TCRs expressed by different subsets of γδ T cells enable them to recognize ligands that are specifically expressed in infected or stressed cells in particular anatomical sites where these cells populate. For example, epidermal intraepithelial Vγ5Vδ1 (DETCs) cells have been shown to carry out distinct functions which are not typical of other γδ T cells, such as production of keratinocyte growth factor, which plays an important role in wound healing. These cells form a dendritic network which is unique among T cells, but similar to that of Langerhans cells, the antigen-presenting cells of the epidermis. In physiological states, DETCs constitute more than 90 % of the epidermal T cells, with virtually no TCR diversity [8].
Vγ6Vδ1 T cells comprise the vast majority of the intraepithelial lymphocytes of the tongue and reproductive tract. These cells seem to play an important role in tissue remodelling at the maternal-fetal interface [9]. Moreover, Vγ6Vδ1 were also shown to mainly produce IL-17 during pulmonary inflammation, thus preventing lung fibrosis [10].
Cells that express the Vγ7 TCRγδ (usually paired with Vδ4 or Vδ5) are typically found as IELs (intestinal epithelial lymphocytes) in gut epithelia and show cytoprotective, immunomodulatory, and antibacterial functions. These protective functions are associated with the production of epithelial cell trophic factors, inflammatory cytokines (such as IL-2 and IFN-γ), and cytotoxic molecules [11].
Cells that express Vγ1 and Vγ4 constitute the major peripheral recirculating γδ T-cell subsets of the blood and lymphatics. Vγ1 cells are capable of killing Listeria-infected macrophages via Fas/Fas ligand [12] and are also shown to promote mouse chronic granulomatous disease [13]. The Vγ4 population tends to be IL-17 biased, whereas the Vγ1 population tends to produce IFN-γ [14].
13.2.2 Human γδ T-Cell Subsets
Human γδ T cells use three main Vδ and at most six Vγ region genes to make their TCRs [3]. Nevertheless, the actual peripheral γδ TCR combinatorial diversity is even more limited because the TCR V region repertoire of human γδ T cells, as in rodents, is highly skewed in particular tissue locations [15].
The two main populations of human γδ T cells constitute the Vδ1 and the Vγ9Vδ2 subsets. Vδ1 T cells are abundant in mucosal tissues, where they are thought to be involved in maintaining epithelial tissue integrity following damage, infection, or transformation [3]. Vγ9Vδ2 T cells dominate (60–95 % of all γδ T cells) in the blood, where they comprise 1–10 % of circulating lymphocytes in healthy adults.
Similarly to mice, the first γδ T cells to emerge in the human fetal thymus, which are Vδ1 T cells, preferentially populate epithelial tissues such as the intestine [16]. Vγ9Vδ2 T cells derive from a subsequent pool of thymic progenitors. By studying γδ T cells from the thymus or peripheral blood of children, it was revealed that the Vγ9Vδ2 pairing makes up only 5 % of γδ thymocytes, indicating selective (chronic) expansion of Vγ9Vδ2 T cells in the periphery [17]. Such extensive peripheral expansion seems to be driven by antigens present in environmental microbes and certain edible plants which stimulate Vγ9Vδ2 T cells during childhood. Of note, this Vγ9Vδ2 pairing is only present in humans and nonhuman primates [3, 18] and therefore has no equivalent in mice.
Vγ9Vδ2 and Vδ1 T-cell subsets differ in several aspects. Most Vγ9Vδ2 T cells display a memory phenotype acquired during perinatal life, whereas Vδ1 T cells are mainly naive in young adults [19]. Vγ9Vδ2 T cells express more cytokines involved in promoting inflammation, such as TNF-α, IFN-γ, and IL-21, and higher levels of CCR5, suggesting that they can home to sites of inflammation [20]. By contrast, Vδ1 T cells express higher levels of L-selectin and CCR7, conferring that they can home to non-inflamed tissues. Furthermore, while Vγ9Vδ2 T cells react against a set of non-peptidic, phosphorylated compounds (“phosphoagonists”), Vδ1 T cells seem to recognize unrelated antigens still poorly defined. In the context of the robust response of Vδ1 T cells to cytomegalovirus (CMV) infection, it was suggested that putative antigens are not virally encoded but instead consist of endogenous stress-induced ligands possibly shared by CMV-infected cells and several colon tumors [21]. Finally, Vγ9Vδ2 cells, but not Vδ1 cells, were recently shown to display (upon activation) several features of professional APCs, namely, the capacities to phagocytize and process antigens; to either present antigens on MHC-II or cross-present antigens on MHC-I; to upregulate CD80, CD86, or CD40; and to activate naive αβ T cells [22, 23]. The APC function of Vγ9Vδ2 T cells adds a new component to the role of γδ T cells as a “bridge” between innate and adaptive immunity.
13.3 γδ T-Cell Activation: TCRγδ Agonists
Immunologists have been searching for TCRγδ ligands for about two decades. However, this has proven to be a very difficult task, likely due to the low affinity interactions that prevent biochemical purification of the putative ligands. An important characteristic of γδ T cells is that they do not recognize classical TCR ligands (peptides derived from processed proteins) and do not depend on MHC-mediated antigen presentation, which markedly distinguishes them from αβ T cells.
It is postulated that γδ T cells recognize a diverse set of “stress-associated” molecules, which may be complexed (or not) with an antigen-presenting element (distinct from classical MHC). As more TCRγδ ligands will become elucidated, it will be interesting to determine whether they comprise molecules whose major function is to regulate immunity (as we conventionally view MHC) or molecules with intrinsic function(s) related to cellular dysregulation, e.g., heat-shock proteins [4]. Below, the authors review the state of the art on the molecular entities suggested to activate γδ T cells in a TCR-dependent manner.
13.3.1 Phosphoagonists (Phosphoantigens)
13.3.1.1 Phosphoagonists Produced by Microorganisms and Eukaryotic Cells
Early in vitro studies indicated that Vγ9Vδ2 T cells strongly react in a non-MHC-restricted fashion to inactivated Mycobacterium tuberculosis and a variety of other microorganisms, including Plasmodium falciparum, Toxoplasma gondii, Yersinia enterocolitica, and Francisella tularensis [24–28]. It was found later that the γδ T-cell-stimulating moiety of microbial extracts was not protein but rather consisted of phosphatase-sensitive low-molecular-weight compounds [28, 29]. Different types of phosphorylated ligands were isolated from Mycobacteria, including four structurally related phosphoesters (so-called TUBag [1–4] 1996) [30]. The other identified phosphate-containing antigens were isopentenyl pyrophosphate (IPP) and its isomer dimethylallyl pyrophosphate (DMAPP). These molecules were collectively termed “phosphoantigens” [30–32].
As a class of compounds, phosphoantigens contain multiple members, either naturally produced or synthetic, able to activate Vγ9Vδ2 T cells within a very large range of affinities [33]. The most potent natural phosphoantigen identified to date is a phosphorylated intermediate of isoprenoid biosynthesis pathway, produced by Eubacteria and Protozoa, but not by eukaryotes, called E-4-hydroxy-3-methylbut-2-enyl-pyrophosphate (HMB-PP, also known as HDMAPP for hydroxy-dimethylallyl pyrophosphate) [28, 34].
The intracellular mechanisms of HMB-PP-mediated Vγ9Vδ2 T-cell activation were previously described [35]. HMB-PP activates MEK/Erk and PI-3K/Akt pathways with similar kinetics to TCR/CD3 cross-linking using OKT3 (anti-CD3ε mAb) and induces an almost identical transcriptional profile associated with γδ T-cell activation, proliferation, and antitumor cytotoxicity [35].
Antibody blocking and gene transfer experiments showed that Vγ9Vδ2 TCR expression is required for cell activation [25, 36]. Nevertheless, it is still controversial if there is a direct interaction between the Vγ9Vδ2 TCR and phosphoantigens – for which the designation “phosphoagonists” may be more appropriate. In particular, while some studies suggested a direct ligation between Vγ9Vδ2 TCR and phosphoagonists [37, 38], all the attempts to co-crystallize phosphoagonists with the Vγ9Vδ2 TCR have not been successful [39].
Very recently, Scotet, Bonneville, and co-workers showed that butyrophilin 3A (CD277/BTN3A) plays a key role in phosphoagonist-induced activation of Vγ9Vδ2 T cells in both tumor and infectious contexts and that CD277-dependent activation is conferred by Vγ9Vδ2 TCR [40]. Their work suggests that phosphoagonist may interact more directly with CD277 than the Vγ9Vδ2 TCR. How Vγ9Vδ2 T cells may detect phosphoagonist-induced changes of CD277 remains to be determined. These changes could be sensed directly by Vγ9Vδ2 TCR; however, the authors failed to demonstrate cognate interactions between recombinant Vγ9Vδ2 TCR and CD277 [40]. Alternatively, CD277 might promote recruitment of other molecules that interact with the Vγ9Vδ2 TCR, such as ecto-F1-ATPase [41, 42].
13.3.1.2 Phosphoagonist Intermediates of Isoprenoid Biosynthetic Pathways
Isoprenoids are essential metabolites, important for cellular and intercellular biology, and are produced by all living organisms. They constitute a diverse structural family, comprising ubiquinones, sterols, terpenes, carotenoids, gibberellins, and taxoids. All these compounds are synthesized through the same precursors, the IPP, and its isomer dimethylallyl pyrophosphate (DMAPP). IPP can be synthesized via two different biosynthetic pathways. Archaebacteria, few Eubacteria, and most eukaryotes synthesize IPP from acetyl CoA through the mevalonate pathway (MVA) [43]. Cyanobacteria, algae, plastids, and most Eubacteria (including M. tuberculosis) produce IPP in a different way, through a carbohydrate-based route referred to as 1-deoxy-d-xylulose-5-phosphate (MEP pathway or DOXP pathway) [44]. Which of these two pathways, MEP or MVA, have evolved first remains unknown, since MEP only exists in bacteria and plastids where it provides most primary isoprenoids instead of the MVA used by Archae [45]. Both pathways can be used simultaneously by some bacterial species, but for different roles, MAP for primary metabolism and MVA for secondary metabolites [46].
Vγ9Vδ2 T cells recognize metabolites of isoprenoid synthesis generated by the MEP pathway in certain pathogenic microorganisms but not by the mevalonate pathway in other bacteria and mammalian cells. HMB-PP has a 1,000-fold stronger stimulating activity of Vγ9Vδ2 T cells than IPP, probably due to its nonhuman origin [28, 32]; this may allow the efficient detection of infected cells producing very small amounts of microbial phosphoantigens, while preventing activation by normal cells that express basal levels of the weak stimulatory mammalian metabolites. Moreover, the high potency of HMB-PP as a stimulator of Vγ9Vδ2 T cells correlates with the γδ T-cell stimulatory activity of the bacteria exploiting the MEP but not the MVA pathway (like Mycobacterium tuberculosis and E. coli) [47]. To a lesser extent, the synthetic bromohydrin pyrophosphate (BrH-PP) is also considered as a strong activator of Vγ9Vδ2 T cells and is frequently used in experimental procedures [33].
In plants and yeast, regulation of the MVA pathway occurs at the HMGR level [48]. High levels of farnesyl pyrophosphate (FPP), sterols, or phenylalanine inhibit HMGR activity. In mammalian cells, the HMGR activity is inhibited by statins [49] and phenylalanine [50] or by a feedback inhibition with aminobisphosphonate-induced FPP accumulation [51]. The HMGR activity and, thus, the whole MVA pathway are increased in various cancer cell types such as leukemia, non-Hodgkin lymphoma (NHL) [52], and mammary and lung adenocarcinoma [53, 54].
13.3.2 Aminobisphosphonates
In 1999, Kunzmann et al. discovered that several patients with multiple myeloma (MM) treated with the well-established osteoporosis inhibitor pamidronic acid (pamidronate) presented significantly high numbers of blood-borne γδ T cells [55]. Later, it was shown that pamidronate activates γδ T cells in vitro to secrete cytokines (IFN-γ), proliferate, and exhibit strong cytotoxicity against various cancer cell lines [37]. Importantly, the bioactivity of aminobisphosphonates like pamidronate required the presence of accessory “antigen-presenting cells” (APCs) treated with this drug prior to the assay with the γδ T cells [36]. A wide variety of tumor cell lines pretreated with aminobisphosphonates could efficiently activate Vγ9Vδ2 T cells to proliferate and produce cytokines in a TCR-dependent manner [56]. Zoledronate and ibandronate are more potent than pamidronate in promoting Vγ9Vδ2 T-cell activation [57].
It is well known that, in order to activate Vγ9Vδ2 T cells, aminobisphosphonates must be internalized and exert a statin-sensitive effect, namely, inhibiting the endogenous MVA pathway [32]. Thus, aminobisphosphonates cause a pharmacological inhibition of the mevalonate pathway in the treated cells leading to IPP accumulation. More precisely, aminobisphosphonates are inhibitors of the farnesyl pyrophosphate synthase (FPPS), an enzyme acting downstream of IPP along the pathway [32]. Of note, non-aminobisphosphonate inhibitors for osteoporosis such as etidronate or clodronate neither inhibit the MVA pathway nor enable Vγ9Vδ2 T-cell activation.
13.3.3 Alkylamines
Similarly to aminobisphosphonates, alkylamines were shown to inhibit FPPS activity. Thus, Vγ9Vδ2 T cells can be activated through accumulation of phosphoagonists in alkylamine-treated cells. Alkylamines are structurally composed of nonphosphate short alkyl chains bearing a terminal amino group. Prototypic bioactive alkylamines are ethylamine and sec-butylamine, present in wine and green tea and produced by certain plants and bacteria. Listeria monocytogenes, Bacteroides fragilis, Proteus morganii, Clostridium perfringens, and Salmonella typhimurium produce alkylamines in concentrations able to activate Vγ9Vδ2 T-cell responses [58]; contrary to phosphoagonists, they only work in the millimolar range (compared to nanomolar to picomolar for phosphoagonists). The activated Vγ9Vδ2 T cells then release abundant Th1-type cytokines and for this reason, it is thought that alkylamine-rich diets may contribute to prevent (Th2-driven) food allergies [49].
13.3.4 Protein Ligands
13.3.4.1 Self-Ligands
Several self-proteins thought to report cellular “stress” have been shown to activate γδ T cells via the TCR [15].
T10/T22
T10 and T22 are murine nonclassical MHC class I molecules expressed by highly activated cells that have been shown to bind specifically to two TCRγδ molecules (G8 and KN6) in surface plasmon resonance experiments [59, 60]. The crystal structures of these murine TCRγδ complexed with T10/T22 have also been solved [60]. So far, these are the only structural evidences for direct binding of TCRγδ to its ligand. Although MHC-I related, T10 and T22 do not present peptides or lipids, being instead recognized as intact proteins via contacts with an extended complementary-determining region (CDR)3 loop of TCRγδ [60–62]. T10-/T22-specific γδ T cells represent 0.4–0.6 % of the peripheral γδ T-cell pool of naive mice [59]; however, this reactivity is not conserved in humans (where T10 and T22 do not exist).
F1-ATPase
The human Vγ9Vδ2 TCR was shown to bind to Ecto-F1-ATPase, a form of the mitochondrial ATP synthase (ATPase) ectopically expressed at the cell membrane. This ligand was identified by screening monoclonal antibodies capable of inhibiting the recognition of tumor cell lines by Vγ9Vδ2 T cells in vitro [41]. F1-ATPase is recognized by Vγ9Vδ2 TCR in a complex with the serum protein apolipoprotein A1 (ApoA-1). These components seem involved in endogenous phosphoantigen presentation, considering the ability of ecto-F1-ATPase to bind and present triphosphoric acid 1-adenosin-5′-yl ester 3-(3-methylbut-3-enyl) ester (ApppI) [63]. ApppI is an intracellular nucleotidic metabolite containing an isopentenyl moiety that accumulates in aminobisphosphonate-treated cells. ApppI can specifically activate Vγ9Vδ2 T cells, but not in its native form; it requires processing by a nucleotidic pyrophosphatase (NPP), which releases IPP and AMP. In this regard, ApppI should represent an inactive storage form of phosphoantigens that can only bind to ecto-F1-ATPase upon cleavage by NPP and generation of IPP [63].
However, the biological relevance of this interaction is still being addressed. It is possible that mitochondrial antigens could be an alerting signal that indicates the status and fate of the cell. On the other hand, the interaction between these molecules could be justified by the specific microbial origin of mitochondria, carrying antigens similar to modern microbes.
ULBP4
The nonclassical MHC class Ib protein, ULBP4, was detected on the cell surface of EBV-infected cells as well as on colon, ovarian, and liver cancer cells, suggesting a role in anti-infection and antitumor immunity. Immobilized soluble ULBP4 was shown to bind directly to soluble Vγ9Vδ2 TCR and to stimulate the activation of Jurkat Vγ9Vδ2 TCR transfectants (lacking NKG2D expression) [64]. Furthermore, ULBP4 ligation induced proliferation, cytokine production, and cytotoxic activity of human ovarian and colonic carcinoma-infiltrating Vγ9Vδ2 T cells in vitro. However, blocking experiments indicated that both Vγ9Vδ2 TCR and NKG2D are involved in ULBP4 recognition [64], raising questions about the hierarchy between NKG2D and Vγ9Vδ2 TCR in γδ T-cell activation and target recognition (Table 13.2).
Table 13.2
Expression of NKG2D in lymphocyte subsets
Cell type | Mouse | Human |
|---|---|---|
NK cells | 100 % | 100 % |
CD8+ T cells | Before activation: absent | Before activation: ≈100 % |
After activation: ≈100 % | After activation: ≈100 % | |
CD4+ T cells | Rare or absent | Normally absent |
γδ T cells | Spleen (Vγ4 and Vγ1): ≈25 % | Blood (Vγ9Vδ2)≈100 % |
IELs (Vγ7): absent | Blood (Vδ1)≈100 % | |
Skin DETCs (Vγ5Vδ1): ≈100 % | IELs (Vδ1)≈100 % |
MICA
Dual recognition of tumors and infected cells is achieved by human Vδ1 cells, as TCR-dependent responses toward both epithelial cell-derived tumors and infected cells have been shown [21]. MICA has been proposed as an important tumor antigen, with recognition of MICA-positive tumor cells by Vδ1 lymphocytes infiltrating colon carcinomas [65–67]. Nevertheless, the very low affinity of MICA-Vδ1TCR interactions estimated by surface plasmon resonance analyses raises doubts about the functional relevance of MICA recognition by Vδ1 TCRs [68].
EPCR
Recently, a human Vγ4Vδ5 clone was shown to directly bind endothelial protein C receptor (EPCR), which allowed γδ T cells to recognize both endothelial cells targeted by CMV and epithelial tumors. EPCR is a major histocompatibility complex-like molecule that binds lipids analogously to the antigen-presenting molecule CD1d [69].
Heat-Shock Proteins (HSP)
Because of their role as sensors during cell stress or transformation, HSP (heat-shock proteins) were initially proposed as antigenic targets for γδ T cells. Some members of HSP were shown to be upregulated on tumors, where γδ T cell had infiltrated, suggesting HSP-65-dependent recognition of tumor cells by Vγ9Vδ2 T lymphocytes [46, 70]. Also, HSP-60 was shown to be recognized by Vγ9Vδ2 T cells [71] and promote their expansion [72].
13.3.4.2 Non-Self-Ligands
Tetanus toxoid, a strong immunogen derived from a protein, the tetanospasmin of Clostridium tetani, was the first defined antigen reported to be capable of stimulating γδ T-cell responses [73, 74]. Others that followed include viral proteins such as glycoprotein I from herpes simplex [75] and staphylococcal enterotoxin A [76]. More recently, the defined mycobacterial protein ESAT-6 was found to stimulate γδ T cells [77], and this may not be the only mycobacterial protein recognized by γδ T cells [78].
13.4 γδ T-Cell Activation: Costimulatory Molecules
T-cell activation depends not only on TCR triggering but also on signals from several additional receptors, commonly referred to as costimulatory molecules. Although these mechanisms have been extensively studied for conventional αβ T cells, they are less well established for γδ T cells [79].
13.4.1 CD27
CD27 is a member from the TNF-receptor superfamily that plays critical roles on γδ Τ-cell activation, particularly in response to viral and tumor challenge [80]. The ligand for CD27 is CD70, and the interaction between these molecules provides a potent second signal for cytokine production, induction of activation markers, and proliferation of primed and unprimed peripheral blood lymphocytes [81].
The authors have shown that the expression levels of CD27 define two stable subsets of γδ T cells in naive C57BL/6 mice [14, 79]. The majority of γδ T cells in the spleen, lymph nodes, and various tissues are CD27+ and secrete IFN-γ upon activation. By contrast, IL-17 is only produced by their CD27− counterparts. Interestingly, these distinct phenotypes are “preprogrammed” in the thymus, as early as in embryonic stages [14, 82]. Moreover, CD27 stimulation (using soluble recombinant CD70) in fetal thymic organ cultures favored the development of IFN−γ+ γδ T cells [14].
In the periphery, CD70-CD27 interactions provide survival and proliferative signals that control TCRγδ-driven activation. Thus, CD27 signalling activates the noncanonical NF-κB pathway and enhances the expression of antiapoptotic and cell cycle-related genes in murine γδ T cells [79, 83, 84].
In humans, an average of 80 % of Vγ9Vδ2 T cells express CD27 [83] including both naive and central memory cells [85]. Upon activation with PMA and ionomycin, the vast majority of CD27+ Vγ9Vδ2 T cells produce IFN-γ, whereas less than 1 % produce IL-17 [83]. A recent work performed by the authors demonstrated that CD70-CD27 interactions enhanced survival and proliferation of phosphoantigen-activated Vγ9Vδ2 T cells and promoted their Th1-like responses (i.e., the secretion of IFN-γ and TNF-α) [83]. Thus, a major role of CD27 costimulation in Vγ9Vδ2 T cells appears to be the protection from activation-induced cell death (AICD) following phosphoantigen-mediated (TCR-dependent) stimulation [83]. Interestingly, CD70 is strongly induced in phosphoantigen-activated Vγ9Vδ2 T cells, which may therefore provide their own CD27 ligands during immune responses.
13.4.2 CD28
CD28, the receptor for B7.1 (CD80) or B7.2 (CD 86), is the primary costimulatory receptor for αβ T cells. CD28 signalling has been shown to produce both qualitative and quantitative changes leading to lower activation thresholds and enhanced αβ T-cell functions. CD28 signalling promotes proliferation, survival, and cytokine production of CD4+ and CD8+ T cells, and such responses are frequently impaired in Cd28 −/− mice [86].
CD28 is upregulated upon activation in murine γδ T cells and it is expressed by 40–60 % of freshly isolated human peripheral blood γδ cells [79, 87]. Although some reports suggested that CD28 costimulation promotes the proliferation of peripheral γδ T cells, other biological processes appeared to be CD28 independent [79].
The authors have recently revisited the role of CD28 costimulation in γδ T-cell activation. It was observed that CD28, constitutively expressed on freshly isolated lymphoid γδ T cells, promoted γδ Τ cell survival and proliferation in both mice and humans. Thus, γδ cell expansion was significantly enhanced by CD28 receptor agonists but abrogated by B7 antibody-mediated blockade [87]. Mechanistically, it was shown that the induction of IL-2 production is a major and specific function of CD28 (but not CD27) costimulation in γδ cells, which are known to strongly benefit from IL-2 signals for their expansion [35, 88]. The fact that γδ cells can produce high levels of IL-2 strictly upon CD28 costimulation defines important rules for their expansion in situ. Of note, CD28-deficient mice displayed reduced (relative to WT controls) numbers of total or activated γδ cells following Plasmodium berghei infection, which was not phenocopied in CD27-deficient animals. This demonstrates that the two costimulatory pathways play independent roles in γδ T-cell activation in vivo [87]. Most importantly, CD28-deficient mice failed to expand both IFN-γ+ and IL-17+ γδ T cells in response to Plasmodium parasites [87], which contrasted with the selective effect of CD27 on IFN-γ-producing γδ cells [84]. Regarding the latter, the authors further showed that CD28 acts nonredundantly and synergistically with CD27 in their activation and expansion following malaria infection [87].
13.4.3 Fc Receptors: CD16
NK cells are able to detect IgG antibody-coated cells through the FcγRIIIA (CD16) cell-surface receptor and to exert antibody-dependent cell cytotoxicity (ADCC) and cytokine production. Specifically, higher cytolytic activity and early IFN-γ production are functional properties of CD56dimCD16+ NK cells [89]. CD16 is coupled to the CD3ς and FcRγ signal transduction proteins bearing ITAMs (immunoreceptor tyrosine-based activation motifs). Besides NK cells, a subset of Vγ9Vδ2 T cells has been shown to express CD16. CD16 upregulation is associated with terminal differentiation into effector cells of both αβ and γδ T cells. Interestingly, Angelini et al. showed that this phenotypic differentiation was associated with decreased Vγ9Vδ2 TCR signalling that paralleled enhanced CD16-mediated T-cell activation [90]. The mechanisms underlying the balanced contribution of TCR vs. CD16 signalling along γδ T-cell functional differentiation remain unclear. Nevertheless, experiments led by Lafont et al. have highlighted the role played by CD16 engagement in γδ T cells. Indeed, cross-linking of CD16 on Vγ9Vδ2 T lymphocytes initiates intracellular signalling events similar, although significantly delayed, to those occurring following TCR activation. Moreover, as observed with the TCR activation process, CD16-triggered TNF-α production can be efficiently inhibited by the coincident ligation of CD94/NKG2A [91].
Recently, the activation of Vγ9Vδ2 T cells with the synthetic phosphoantigen BrH-PP was shown to improve the efficacy of cancer immunotherapy by the therapeutic mAb rituximab (RTX). Thus, combination of BrH-PP with RTX increased Vγ9Vδ2 T-cell binding and ADCC activity against CD20+ lymphoma cells in vitro. Moreover, a regimen combining RTX, BrH-PP, and IL-2 activated Vγ9Vδ2 T lymphocytes and enhanced B-cell depletion from blood and lymph nodes of cynomolgus macaques [92].
13.5 γδ T-Cell Activation via Natural Killer Receptors (NKRs)
13.5.1 NKG2D
Natural killer group 2 member D (NKG2D) is an activating C-type lectin receptor expressed on the surface of NK cells, CD8+ T cells, and γδ T cells [93] (Table 13.2). NKG2D activation is best described in NK cells, where its cross-linking (on murine NK cells) was shown to trigger several effector mechanisms, such as Th1 cytokine production (IFN-γ, GM-CSF, TNF-α) and the release of cytotoxic granules [94, 95].
NKG2D itself does not possess signalling capacity. In humans, NKG2D exists on the cell surface complexed with the DAP10 adaptor protein that contains a YxxM motif which, upon tyrosine phosphorylation, couples the receptor complex to the PI3K/Grb2-Vav pathway [96, 97]. Murine NKG2D is encoded by two splice variants [98]. The long isoform (mNKG2D-L) associates only with DAP10, whereas the short isoform (mNKG2D-S) associates with DAP10 or DAP12 [98, 99].
Several mechanisms are known to regulate the cell-surface expression of the NKG2D receptor, including the differential action of particular cytokines. Thus, TGF-β1 [100–102] and IL-21 [103] lead to downregulation of NKG2D expression on NK and CD8+ T cells. By contrast, IL-2 and IL-15 signals increase NKG2D surface expression [104, 105] by upregulating DAP10 mRNA and protein synthesis. Interestingly, it was shown that TCR ligation in CD8+ T cells also upregulates NKG2D/DAP10 cell-surface expression [106], which may underlie a costimulatory function for NKG2D in CD8+ T cells.
The role of NKG2D in T cells remains controversial, as some authors argue that NKG2D has solely a costimulatory function, whereas others defend that NKG2D signals can activate T cells in the absence of TCR engagement. Thus, for human CD8+ T cells, various reports showed that NKG2D-DAP10 can mediate cytolysis independent of TCR engagement when cells are exposed to IL-15 or high-dose IL-2 [105, 107–109]. Specifically for γδ T cells, some studies reported the ability of Vγ9Vδ2 T cells to trigger effector responses through NKG2D stimulation alone [110, 111]. However, others have failed to show any Vγ9Vδ2 T-cell NKG2D-induced activation without coincident TCR stimulation [112, 113]. In particular, it was recently shown that NKG2D triggering per se could not produce calcium fluxes in γδ T cells, but its co-engagement with TCR/CD3 significantly augmented the intensity of calcium responses, which also translated into enhanced cytotoxicity (while not affecting IFN-γ production) [113].
The ligands for NKG2D belong to the MHC class Ib protein family (also known as nonclassical MHC), which are usually upregulated on transformed, stressed, or infected cells. The MHC class Ib molecules are structurally related to class Ia proteins in that they show typical (α1–α2) MHC fold on a single polypeptide, which, in the case of Ib, does not obligatorily paired with β2-microglobulin. Furthermore, although many MHC Ib genes are located in the MHC locus, they tend to be oligomorphic, with few alleles present in the population (with the notable exception of MICA/B), which markedly contrasts with the extensive polymorphism of class Ia [114]. MHC class Ib molecules can work as ligands for particular types of TCRs or NK receptors, most notably NKG2D [114].
Mouse NKG2D binds to retinoic acid early transcript (Rae1), histocompatibility antigen 60 (H60), and murine UL16-binding protein-like transcript 1 (MULT1) (Fig. 13.1). Human NKG2D binds to MHC I chain-related (MIC) peptides A and B (MICA and MICB) and to UL16-binding proteins (ULBP, members 1–6) (Fig. 13.1) [114, 115]. MICA/B, ULBP4, H60, and MULT1 are transmembrane proteins, while ULBP1, ULBP2, ULBP3, ULBP5, and ULBP6 and Rae1 localize to the cell surface using glycosylphosphatidylinositol (GPI) linkages [93, 115]. None of the NKG2D ligands bind to peptide or lipid antigens but rather interact directly with the receptor. In addition, NKG2D ligands do not associate with β2-microglobulin [93] in contrast to some other members of the MHC class Ib family (e.g., HLA-G or CD1d).
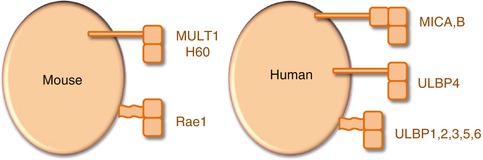
Fig. 13.1
Mouse and human NKG2D ligands. All NKG2D ligands have α1 and α2 domains with structural homology to MHC class I, and MICA and MICB have also a α3 domain. By contrast with MHC class I, none of the NKG2D ligands associates with β2-microglobulin or binds peptides. MULT1, H60, MICA/B, and ULBP4 are transmembrane-anchored type I glycoproteins, whereas Rae1 and ULBP1, ULBP2, ULBP3, ULBP5, and ULBP6 bind to cell membrane by a GPI anchor
NKG2D ligands are usually induced by a variety of signals associated with cellular stress, namely, oxidative stress, ionizing radiation, DNA-damaging agents, viral infections, and intracellular bacterial infections [116]. Nonetheless, the various NKG2D ligands have distinct patterns of expression, indicating that they cannot be considered simply redundant in function.
Despite the marked differences in their amino acid sequences, the different ligands interact with NKG2D in similar fashion, and the receptor does not seem to undergo marked conformational changes to accommodate different ligands [117]. So far, there is no evidence that the different ligands induce qualitatively distinct biological effects in responding cells, though this remains a possibility. Minimally, the various ligands would be predicted to differ quantitatively in their effects based on the marked differences in their affinity for NKG2D. At present, the relevance of such differences has not been documented.
The murine ligands Rae1 and H60 are rare in healthy adult tissues, but their transcription is strongly induced in keratinocytes after their exposure to carcinogens in vivo [118], and they are overexpressed in the cutaneous papillomas and carcinomas that subsequently develop, as well as in various tumor cell lines [98, 119]. The expression of Rae1 or H60 by target cells was shown to enhance cytolysis and the production of IFN-γ by CTLs [120] and γδ T cells [118] leading to tumor rejection in vivo. Moreover, transduction of Rae1, H60, or MULT1 into NK-cell-resistant target cells made them susceptible to NK-cell-mediated killing and stimulated IFN-γ secretion [120, 121].
In contrast to other mouse ligands (Rae1 and H60), MULT1 is expressed at marked levels by various normal cells at the mRNA level [122], but cell-surface expression is low or has not been documented. For example, C57BL/6 thymocytes contain high levels of Mult1 mRNA but stain poorly with NKG2D tetramers [123]. However, MULT1 is expressed at functional levels on the cell surface of numerous tumor cell lines, indicating that these molecules might be regulated at a level other than transcription [123].
The human MICA and MICB proteins show restricted and low expression in healthy tissues but are strongly induced by cellular stress (including heat shock) and transformation. In addition, they accumulate in various tumor cell lines, particularly those of epithelial origin [66, 124]. Upregulation of MICA and MICB expression by these cells seems to result from activation of heat-shock transcription elements in the promoters of the corresponding genes, an event known to accompany transformation [66]. Interestingly, heat-shock elements have not been implicated in regulating the expression of Rae1, H60, MULT1, or ULBPs. Atypically for MHC Ib molecules, the MIC genes are highly polymorphic consisting of 61 MICA and 30 MICB alleles [93].
Whereas the membrane-bound form of MICA provides stimulatory signals to killer lymphocytes, soluble forms that shed from the cell surface may downregulate surface NKG2D and impair tumor cytolysis, constituting an important immune evasion mechanism [125, 126]. Moreover, NKG2D ligands can be expressed by tumor-released exosomes [127] that promote downregulation of surface NKG2D expression by NK and CD8+ T cells. Interestingly, a similar phenomenon occurs in human placenta to avoid immunosuppression during pregnancy [128].
Distantly related to the MIC proteins are the members of the ULBP family. In contrast with Rae1 or MICA, ULBPs are expressed at significant levels in a wide range of healthy tissues and cell lines of both epithelial and non-epithelial origin [129, 130]. Ectopic expression of ULBP1 or ULBP2 on murine EL4 or RMA tumor cells elicits potent antitumor responses in syngeneic C57BL/6 and SCID mice, recruiting NK, NKT, and T cells to the tumor [131]. Similarly, tumor cells that are insensitive to NK cells can be lysed effectively when transfected with ULBPs [132]. Moreover, tumor cell susceptibility to current first-line treatment to NHL, rituximab (anti-CD20 mAb), was shown to greatly depend on ULBP1–ULBP3 expression [133].
We have demonstrated that ULBP1 is a nonredundant determinant of hematological tumor susceptibility to Vγ9Vδ2 T cells [134]. By using loss- and gain-of-function studies, the authors have shown that ULBP1 expression on leukemia and lymphoma cell lines is required and sufficient for Vγ9Vδ2 T-cell recognition [134]. Moreover, leukemic B cells were also shown to express ULBP3 that is recognized by Vδ1 T cells, the other major subset of human γδ T cells [135].
Furthermore, epithelial tumors, such as ovarian and colon carcinomas, which express low or undetectable levels of ULBP1 [110], seem to rely on ULBP4 for Vγ9Vδ2 T-cell recognition [64].
Cancer cells can also shed proteins of the ULBP family. ULBP2 is secreted both from tumor cell lines and primary tumor cells from patients and sera-soluble ULBP2 was shown to have poor prognostic value in melanoma patients [136]. Other studies also correlate NKG2D ligand expression with cancer clinical prognosis; for example, loss of ULBP1 in hepatocellular carcinoma correlates with tumor progression and early recurrence [137], whereas expression of MICA/B and ULBP2 in breast cancer is an independent prognostic parameter for relapse-free period [138].
The expression of human NKG2D ligands seems to be modulated by proteasome regulation. For example, in head and neck squamous cell carcinoma (HNSCC), bortezomib (an approved drug for treatment for plasma cell myeloma) and other proteasome inhibitors with distinct mechanisms of action dramatically and specifically upregulated ULBP1mRNA and cell-surface protein expression. In different types of tumors, such as hepatocellular carcinoma, low-dose proteasome inhibitor drugs caused upregulation of MICA and MICB, but not ULBP1-3 [139]. In contrast, other reports showed that several proteasome inhibitor drugs increased ULBP2 levels on Jurkat surface T cells, whereas MICA, MICB, and ULBP1, ULBP3, and ULBP4 were not affected [140].
Moreover, both murine and human non-tumor cell lines may upregulate NKG2D ligands in response to DNA-damaging agents and DNA synthesis inhibitors. Activation of the DNA damage pathway is frequently activated in tumor cell lines, possibly due to the greater genomic instability of these cells compared with transformed cells [116].
Other mechanisms of NKG2D ligand expression regulation include differences in promoter sequences of the several ligands [141]; cytokine treatment, for example, TGF-β decreased transcription of MICA, ULBP2, and ULBP4 in human gliomas [142, 143] and IFN-γ decreased MICA message levels in melanoma [144]; and induction of p53, which lead to upregulation of ULBP1 and ULBP2 at the tumor cell surface [145].
An open question in the field is why there are so many ligands for the NKG2D receptor. It is possible that the several ligands stimulate NKG2D positive cells to respond to different forms of stress because they are capable of being expressed independent of each other [129, 130, 141] and because they engage NKG2D with different affinities, suggesting that NKG2D ligands may not be functionally equivalent. In any instance, NKG2D is clearly a key determinant of tumor immunosurveillance, since NKG2D-deficient mice show increased growth of epithelial and lymphoid tumors in two transgenic models of de novo tumorigenesis [146].
13.5.2 NKG2A
As previously shown for NK cells, most human Vγ9Vδ2 T cells express several inhibitory NK receptors, including killer Ig-like receptors (KIR), leukocyte Ig-like receptors (LIRs), and lectin-like receptors, such as the NKG2A/CD94 heterodimer.
The NKG2A/CD94 heterodimer is regarded as a crucial complex molecule for the inhibition of γδ T-cell responses [147]. Most of these inhibitory NKRs decrease the killing of target cells expressing high levels of either classical or nonclassical MHC molecules. Due to the broad cellular distribution of some Vγ9Vδ2 TCR agonists such as IPP, which are upregulated on transformed cells, MHC class I-specific inhibitory NKR may selectively downregulate recognition of healthy cells by Vγ9Vδ2 CTL [118, 120, 148]. Accordingly, masking of inhibitory NKRs increases Vγ9Vδ2 T-cell killing of several hematopoietic and non-hematopoietic tumors [149].
13.5.3 Natural Cytotoxicity Receptors (NCRs)
Although TCR and NKG2D play central roles in the activation of γδ T cells, their response to tumors may involve other receptors, such as natural cytotoxicity receptors (NCRs), including the activating receptors NKp30 [150], NKp44 [151, 152], and NKp46 [153, 154].
NKp30 is encoded on chromosome 6 and has no homology with NKp44 and NKp46, which are encoded on chromosomes 6 and 9, respectively [150]. Notably, NKp30 is a pseudogene in mice, with the exception of the wild strain Mus caroli [155]. A functional but low level of NKp30 protein is expressed in resting peripheral chimpanzee NK cells [156]. Several studies have shown that NKp30 is a major activating receptor involved in tumor cell lysis by NK cells. IL-2 [157] and IL-21 [103] induce NKp30 upregulation, whereas TGF-β downregulates NKp30, leading to impaired NK cytotoxicity [158]. Additionally, an NKp30-dull phenotype was shown to be acquired during leukemia development in acute myeloid leukemia (AML) [158, 159] and breast cancer [160] patients. This downregulation is possibly a mechanism of escape from innate immunity.
A recent study conducted by the authors, demonstrated that human Vδ1 T cells can be selectively induced to express NKp30, NKp44, and NKp46 [161]. Importantly, specific gain-of-function and loss-of-function experiments showed that NKp30 makes the most important contribution to TCR-independent leukemia cell recognition. Moreover, the Vδ1 NKp30+ subset is able to target primary hematological tumors highly resistant to fully activated Vγ9Vδ2 PBLs [161].
Several groups have shown the constitutive expression of NKp30 ligands on tumor cells by assessing the binding of soluble NKp30 [162]. However, only one ligand (B7-H6) was demonstrated to be clearly involved in NKp30-mediated tumor cell recognition [163]. B7-H6 is a surface protein similar to other members of the B7 family. In contrast to B7.1 and B7.2, that recognize both CD28 and CTLA-4, B7-H6 is not promiscuous, since it does not bind to any other CD28 family members or other NCRs [163]. Similar to NKp30, but in contrast to other B7 members, a functional B7-H6 gene is missing in Mus musculus.
B7-H6 transcripts have not been detected in most normal adult tissues, consistent with the absence of the protein on circulating cells, isolated from healthy individuals. In contrast, B7-H6 surface expression is observed in a restricted panel of tumor cell lines from various origins including lymphoma, leukemia, melanoma, and carcinoma as well as on primary tumor blood cells [163]. The pattern of B7-H6 expression, which appears so far to be limited to tumor cells, is another example of stress-induced self-recognition by NK cells [164]. However, in pilot experiments, treatment of some NKp30 ligand-negative tumor cells with a panel of DNA-damaging agents had no major effect on B7-H6 expression.
NKp44 is a type I transmembrane protein non-covalently associated in the plasma membrane with a disulfide homodimer of DAP12 (a transmembrane accessory protein that contains an ITAM, which provides intracellular activation signals) [151, 152]. The NKp44 molecule is expressed on the surface of IL-2 stimulated, but not on resting human NK cells, and therefore is referred to as an activation-induced triggering receptor [152]. Anti-NKp44 mAb can reduce NK-cell cytotoxicity toward certain tumor target cells, thereby indicating that these targets express the appropriate ligands for the receptor [151]. However, the identity of NKp44 ligands on tumors is currently unknown.
NKp44 seems to be involved in Vγ9Vδ2 cytotoxicity against MM cell lines lacking expression of NKG2D ligands. However, the percentage of NKp44+ γδ T cells in culture was very low [165], thus raising the question about the biological importance of NKp44 expression on Vγ9Vδ2 T cells. Nonetheless, it seems like NKp44 is important for Vδ1+ γδ T cells, as gain-of-function and loss-of-function experiments demonstrate that NKp44 is also a functional receptor in activated Vδ1+ T cells and mediates tumor cell killing [161]. Importantly, a synergistic effect between NKp30 and NKp44 (with no additional effect of NKp46) was observed [161]. The authors are currently exploiting the potential of NCR+ Vδ1+ T cells in cancer immunotherapy.
13.5.4 DNAM-1
Another important NK receptor is DNAX accessory molecule-1 (DNAM-1 or CD266), a transmembrane glycoprotein that associates with LFA-1. Its ligands include PVR and Nectin-2. In NK cells, DNAM-1 has a role in tumor cell recognition together with NCRs and to a lesser extent with NKG2D [166]. Decreased expression of DNAM-1 has been observed in NK cells from AML patients [158, 167]. In mouse, DNAM-1 is a crucial component of T-cell-mediated immunological surveillance and partially contributes to NK-mediated lymphoma rejection [168].
Importantly, the human Vγ9Vδ2 T-cell subset expresses DNAM-1, and upon recognition of ligands expressed by hepatocellular carcinoma cells, DNAM-1 signals were shown to increase Vγ9Vδ2 cell cytotoxicity and IFN-γ secretion [169]. Furthermore, a recent report demonstrated that Vγ9Vδ2 T cells efficiently killed autologous AML blasts dependent on DNAM-1 and TCR signals. The DNAM-1 ligands, PVR and Nectin-2, were expressed by the targeted AML blasts [170].
13.6 Tumor Cell Recognition by γδ T Cells: TCRs Versus NKRs
Studies on hematological tumors have highlighted the major role played by activating NKRs in tumor cell recognition by human γδ T cells. This was observed for both Vγ9Vδ2+ and Vδ1+NKp30+ T-cell subsets, in which NKG2D and/or NKp30, rather than the respective TCRs, mediated leukemia/lymphoma cell recognition [134, 161].
Some other groups have suggested that γδ T cells recognize tumor targets through TCR interactions with self-ligands overexpressed by tumor cells and simply use NKR signals to fine-tune their activation threshold (reviewed in [5, 171–173]). In this scenario, TCR-mediated activity would be tightly regulated by an interplay between activating and inhibitory NKRs [171].
Building on these considerations, the authors’ current working model includes two stages of γδ T-cell activation/differentiation and tumor cell recognition (Fig. 13.2). First, γδ cells are potently activated by (mostly unknown) TCRγδ ligands in the presence of IL-2. This, which can be achieved for Vγ9Vδ2 cells using (microbial or synthetic) phosphoagonists (plus IL-2), endows them with potent cytolytic (and cytokine-secreting) function but requires a subsequent phase of target identification, namely, for discrimination between tumor and healthy cells. We propose this is mainly determined by activating NKRs that bind stress-inducible proteins which selectively accumulate on the surface of tumor cells. Of note, the segregation of these two processes (activation vs. tumor cell recognition) in experimental systems requires pre-activation of γδ T cells (through the TCR) before testing them against tumor targets. More importantly, we believe the integration of these two phases will be the key for success of γδ cell-based protocols in future cancer clinical trials.
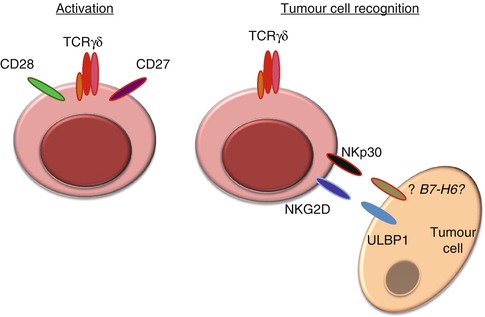
Fig. 13.2
Tumor cell targeting by γδ T cells is two-step process of activation and subsequent tumor cell recognition. γδ T-cell activation requires TCRγδ signalling plus costimulation (CD27 and CD28), whereas tumor cell recognition involves natural killer receptors such as NKG2D and NKp30 (and DNAM-1) that bind counter-ligands (over)expressed on tumor cells
13.7 γδ T-Cell Responses to Tumors
13.7.1 Antitumor Properties
γδ T cells can kill transformed cells, through pathways that involve the engagement of death-inducing receptors, such as CD95 (also known as FAS) and TNF-related apoptosis-inducing ligand receptors (TRAILR), and the release of cytotoxic effector molecules such as perforin and granzymes [173]. Murine IELs, activated DETCs, and human Vγ9Vδ2 cells primarily express granzymes A and B at levels substantially higher than conventional CD8+ T cells. Moreover, a significant fraction of Vγ9Vδ2 cells express intermediate levels of CD16 and thus γδ T cells can improve antibody-dependent cell-mediated cytotoxicity (ADCC) [174].
The importance of murine γδ T cells in tumor immunosurveillance was first described in 2001 by a seminal paper from the Hayday lab. They showed that γδ-deficient mice were highly susceptible to multiple regimens of cutaneous carcinogenesis. Moreover, they observed that the γδ T-cell response in WT mice was determined by NKG2D recognition of Rae1 and H60 molecules, expressed by skin tumor cells. This work further revealed that γδ T cells not only inhibited the early stages of papillomas development but also limited their progression to carcinomas [118].
In the murine B16 melanoma model, γδ T cells were shown to infiltrate tumor lesions already at day 3 posttransplantation and to provide a critical early source of IFN-γ [175]. By using bone marrow chimeras and fetal liver reconstitution experiments, the authors showed that IFN-γ production by γδ T cells seems to be required to control the growth of both MCA-induced tumors and B16 melanoma tumors. This ability of γδ T cells to produce IFN-γ was crucial for the subsequent αβ T-cell activation and differentiation. Thus, depletion of γδ T cells resulted in significantly reduced IFN-γ production by both CD4+ and CD8+ T cells upon challenge with tumor lysates [175]. The direct comparison of protective properties of γδ T cells and αβ T cells was addressed in other chemical carcinogen-induced tumors, namely, squamous cell carcinoma [176]. While papilloma development was comparable in WT and Tcrb −/− mice, it was highly accelerated in Tcrd −/− and in the double-knockout mice, Tcrb −/− d −/−. This study revealed that γδ T cells are strongly protective, whereas the contribution of αβ T cells for tumor progression control is more modest [176].
Subsequent studies also using carcinogen-induced skin tumors reinforced the nonredundant antitumoral role of γδ T cells [177–179]. Moreover, by backcrossing Tcrd –/– mice with TRAMP (transgenic adenocarcinoma mouse prostate cancer) mice, Liu and colleagues showed that γδ T cells limit the development and progression of spontaneously arising mouse prostate cancer [180]. The authors also assessed the possibility of developing an adoptive cell therapy, by treating TRAMP-C2 subcutaneous tumor-bearing mice, with adoptively transferred γδ T cells. Treated mice with supraphysiological numbers of WT γδ T cells develop measurably less disease compared with untreated mice [180].
γδ T cells were also characterized as prototypic antitumor mediators in B-cell lymphomas. Peng and colleagues showed that B-cell lymphomas arose with higher frequency in Fas mutant lpr mice that were additionally deficient for γδ T cells [181]. Moreover, γδ T cells were present in great numbers around B cell tumor masses in the spleens of pfp –/– mice [182]. Also, in this work, both γδ T cells and NK cells were shown to display potent cytotoxicity against spontaneously arising MHC class I-deficient B cell lymphomas.
Studies in mice (Table 13.3) have thus provided important clues to the physiological roles of γδ T cells, but owing to the differences between mouse and human γδ T-cell subsets, these studies have not generally predicted the behavior of human γδ T cells [5].
Table 13.3
Mouse tumor models implicating γδ T cells in tumor immunosurveillance
Spontaneous tumors | Chemical carcinogen-induced tumors | Transplantable tumor cell lines | Tumor type | Reference |
|---|---|---|---|---|
MCA, DMBA+TPA | PDV | Skin fibrosarcoma | [118] | |
Squamous cell carcinoma | ||||
MCA | B16-F0 | Skin fibrosarcoma | [175] | |
Squamous cell carcinoma | ||||
DMBA+ | Squamous cell carcinoma | [176] | ||
TPA | ||||
b2m −/− | Spontaneous B-cell lymphomas | [182] | ||
pfn −/− | ||||
TRAMP × Tcrd −/− | Prostate carcinoma | [180] | ||
DMBA + TPA | Squamous cell carcinoma | [177] |
This notwithstanding, both main subsets of human γδ T cells, Vγ9Vδ2 and Vδ1 cells, have been shown to lyse a broad range of tumor cell lines in vitro. The Vγ9Vδ2+ subset has been more widely studied than the Vδ1 subset, probably due to the easiness of isolation, as they comprise most of the γδ-PBLs. They have been shown to display potent cytotoxicity toward several cell lines of different origins, including breast cancer [183], colon and nasopharyngeal carcinomas [184], melanoma [185], pancreatic adenocarcinomas [185], and particularly a large number of hematopoietic cell-derived tumors [186, 187], including Daudi cell line derived from Burkitt’s lymphoma [48, 188–190], and recently also toward cancer stem cells [191, 192]. However, the frequency of Vδ2 cells within lymphocytes infiltrating solid tumors is generally low, even within Vγ9Vδ2-suscepible tumors such as renal and colon carcinomas [184, 193].
Another important antitumor effect is the induction of IFN-γ-producing Vγ9Vδ2 T cells in vivo. Multiple antitumor effects have been attributed to IFN-γ, including direct inhibition of tumor growth or more indirect effects such as the upregulation of MHC class I molecules and blocking of angiogenesis [194]. Interestingly, a significant negative correlation between the serum levels of the angiogenic factors VEGF (vascular endothelial growth factor) and IFN-γ was found in cancer patients treated with aminobisphosphonates [195].
Conventional mouse models cannot be used to explore the possible antitumor activity of Vγ9Vδ2 cells in vivo, due to the lack of homologous TCR and thus the reactivity to phosphoantigens. However, xenogeneic immune deficiency (SCID) mouse models of human tumors have been established and revealed the efficacy of Vγ9Vδ2 T cells against several human tumors in vivo [35, 185, 196–202]. Pre-activated adoptively transferred human Vγ9Vδ2 T cells localized to tumors [197], increased survival, and inhibited tumor growth [35, 185, 197, 199, 201]. Vγ9Vδ2 T cells are also active against freshly isolated tumor cells from patients with follicular B-cell lymphoma or B-cell chronic lymphocytic leukemia (B-CLL) [203]. Similarly, a high survival rate is obtained when Vγ9Vδ2 TCR+ tumor-infiltrating lymphocyte (TILs) (expanded from human colorectal tumors in vitro) are transferred into Daudi cell-bearing BALB/c nude mice compared with the transfer of αβ TCR+ TILs or mice without treatment [204].
Although less studied, Vδ1 T cells are also promising targets for cancer immunotherapy. Vδ1 tumor-infiltrating lymphocytes from colorectal cancer were shown to lyse autologous and allogeneic colorectal, renal, and pancreatic tumor cell lines [205]. Moreover, circulating Vδ1 cells from chronic lymphocytic leukemia patients were able to lyse B-CLL cells expressing ULBP3 [206]. By contrast, with their Vγ9Vδ2 counterparts, Vδ1 cells are quite frequent within T cells infiltrating solid tumors [193, 205, 207, 208].
The authors have also recently demonstrated that Vδ1 antitumor properties can be enhanced by their culture in the presence of PHA and IL-2 [161]. Fully activated Vδ1 cells display stronger cytotoxicity against B-CLL cells than the corresponding Vδ9Vδ2 counterparts, which was attributed to the selective induction of NCR expression in Vδ1 cells [161].
Interestingly, Vδ1 cells share reactivity toward CMV-infected cells and tumor intestinal epithelial cells [21]. This dual recognition also seems to be a characteristic of the Vγ4Vδ5 clone [69]. Willcox and colleagues demonstrated that Vγ4Vδ5 TCR binds directly to EPCR (endothelial protein C receptor) and that is expressed in both endothelial cells targeted by cytomegalovirus and epithelial tumors [69].
13.7.2 Pro-tumor Properties
The potent antitumoral properties of γδ T cells have been widely shown for more than 15 years. This notwithstanding, some recent studies imply a pro-tumorigenic role for γδ T cells, e.g., γδ T-cell depletion reduced papilloma incidence [209] and breast tumor-infiltrating γδ T cells suppressed naive and effector T-cell responses and blocked maturation and function of dendritic cells [210]. Moreover, intratumoral γδ T cells represented the most significant independent prognostic factor for assessing the severity of breast cancer compared with the other known factors. Intratumoral γδ T cells were positively correlated with FOXP3+ regulatory T cells but negatively correlated with cytotoxic CD8+ T cells in breast cancer tissues [211].
Peng and colleagues have shown that human Vδ1 cells derived from breast cancer biopsies inhibited the maturation and function of dendritic cells and suppressed proliferation and IL-2 production of CD4+ T cells in vitro [210]. Thus, a pro-tumor role of γδ T cells may be linked to immunosuppressive functions that need to be further characterized.
Alternatively, the controversial pro-tumor function of γδ T cells may rely on their production of IL-17, based on a study that showed that murine IL-17-producing γδ T cells promoted tumor growth in a murine fibrosarcoma tumor model [212]. However, murine IL-17-producing γδ T cells were reported to be necessary for BCG treatment of bladder cancer [213] and for chemotherapeutic efficacy in subcutaneous tumor models [214]. Actually, the role of IL-17 in tumor surveillance is itself paradoxical. IL-17 production has been associated with enhanced tumor development/ progression in murine models of intestinal [215], skin [216], bladder [217], and ovarian carcinoma [218]; but, by contrast, IL-17-deficient mice were more susceptible to the development of lung melanoma [219] and lung metastasis [220].
A recent work performed by the authors suggests that γδ T cells promote tumor progression in a mouse model of ovarian cancer (unpublished data). The authors observed that γδ-deficient mice displayed decreased tumor burden compared with wild-type mice. Interestingly, a selective expansion of IL-17-producing γδ T cells in the peritoneal cavity of tumor-bearing mice was observed; therefore, the authors are investigating if γδ T cells promote ID8 tumor progression through the production of IL-17.
Several functions of IL-17 in the tumor microenvironment seem to contribute to tumor progression. Apart from a minor direct effect on the proliferation and survival of tumor cells (as not all tumor cells express the IL-17 receptor and respond to IL-17), the major pro-tumor function of IL-17 in inflammation-associated cancer cells seems to rely on its proangiogenic properties on the surrounding endothelial cells and fibroblasts [221]. By acting on stromal cells and fibroblasts, IL-17 induces a wide range of angiogenic mediators [222, 223], including VEGF, which markedly promotes inflammatory and tumor angiogenesis.
A more detailed characterization of γδ-TILs, in a wider set of preclinical tumor models, is required to clarify the role of IL-17-producing γδ T cells in tumor immunosurveillance. This should take into account the two functional γδ T-cell subsets recently identified: CD27+ γδ T cells produce IFN-γ but no IL-17, whereas IL-17 production is restricted to CD27− γδ T cells [14].
13.8 γδ T-Cell Modulation in Cancer Clinical Trials
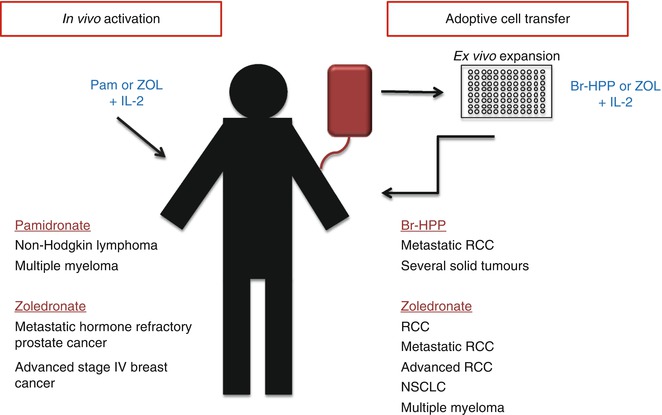
Several features of γδ T cells make them attractive targets for cancer immunotherapy: abundant IFN-γ secretion; potent, broad, and MHC-unrestricted cytotoxicity; and the availability of clinical grade agonists for Vγ9Vδ2 T cells. Vγ9Vδ2 T cells can be directly activated in vivo with TCR agonists or can be expanded in vitro and then reinfused into patients (adoptive cell therapy) [224] (Fig. 13.3). Clinical grade agonists used so far include the synthetic phosphoagonist bromohydrin pyrophosphate (BrH-PP) and the aminobisphosphonates pamidronate and zoledronate. In most clinical trials, recombinant IL-2 (rIL-2; a fundamental cytokine for γδ T-cell expansion) was used in combination with TCR agonists (Table 13.4).
 Novel Strategy of Cancer Immunotherapy: Spiraling Up
Novel Strategy of Cancer Immunotherapy: Spiraling Up
 Dendritic Cell Vaccines for Cancer Therapy: Fundamentals and Clinical Trials
Psychoneuroendocrinoimmunotherapy of Cancer
Dendritic Cell Vaccines for Cancer Therapy: Fundamentals and Clinical Trials
Psychoneuroendocrinoimmunotherapy of Cancer
 Toll-Like Receptor Pathway and its Targeting in Treatment of Cancers
Toll-Like Receptor Pathway and its Targeting in Treatment of Cancers
 Tumor Antigen and Epitope Identification for Preclinical and Clinical Evaluation
Tumor Antigen and Epitope Identification for Preclinical and Clinical Evaluation
 Photodynamic Therapy and Antitumor Immune Response
Photodynamic Therapy and Antitumor Immune Response




Related posts:
Novel Strategy of Cancer Immunotherapy: Spiraling Up
Dendritic Cell Vaccines for Cancer Therapy: Fundamentals and Clinical Trials
Psychoneuroendocrinoimmunotherapy of Cancer
Toll-Like Receptor Pathway and its Targeting in Treatment of Cancers
Tumor Antigen and Epitope Identification for Preclinical and Clinical Evaluation
Photodynamic Therapy and Antitumor Immune Response
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
