Fig. 5.1
Chimeric structure of rituximab. Rituximab is a chimeric anti-CD20 monoclonal antibody containing human IgG1 heavy chain and human kappa light chain constant regions and murine immunoglobulin heavy and light chain variable regions. The molecular formula is C6426 H9900 N1700 O2008 S44 and the molecular weight is 144,510 daltons. CDR complementarity determining region, V H variable regions of heavy chains, V L variable regions of light chains, C H constant regions of heavy chains, C L constant regions of light chains, C1q complement component 1, q subcomponent
5.2.1.2 CD20 Antigen
The CD20 antigen is a 33- to 37-kDa non-glycosylated phosphoprotein with four transmembrane-spanning regions. Although the physiological functions of CD20 have yet to be clarified, it is thought to play an important role in B-cell activation, differentiation, proliferation, and signal transduction. CD20 is expressed on the surface of normal precursor and mature B cells, but not normal B stem cells or plasma cells [5, 6].
5.2.1.3 Favorable Features of CD20 as a Target Antigen
The CD20 antigen is highly expressed on the cellular surface membrane of most B-lymphocyte tumors making it a rational target in the treatment of CLL and B-cell NHLs. CD20 is stable on the membrane of the B cells and is not internalized following the binding of rituximab. It is also not shed or secreted into the circulation in normal individuals resulting in no detectable soluble CD20 serum levels, which might block targeting of the antibody to B-lymphocyte tumors. Since CD20 is not expressed on the normal B stem cells or plasma cells, they are spared from the direct antibody effects resulting in normal mature B cells that can proliferate even after rituximab-induced depletion of B cells. CD20 is not expressed on non-hematopoietic tissues where it could interfere with the antibody [3, 5, 6].
5.2.2 Mechanism of Action of Rituximab
Although the exact or predominant mechanism of how rituximab binding to CD20 results in cytotoxicity has yet to be completely clarified, the proposed mechanisms of action of rituximab include ADCC, CDC, and induction of apoptosis.
5.2.2.1 ADCC
ADCC is mediated by immune effector cells binding to rituximab-coated B cells. This requires rituximab to bind through the Fc region to Fc receptors for IgG (FcγRs) on immune effector cells, such as natural killer (NK) cells, monocytes, macrophages, and granulocytes [3]. ADCC is better mediated through the IgG1 constant region, and polymorphisms in the genes encoding FcγRs may affect the response to rituximab therapy [5].
5.2.2.2 CDC
Rituximab can recruit components of the complement cascade through the Fc region. The first step is the binding of C1q components to the Fc region. C1q activation triggers the classical complement cascade, which leads to the generation of the membrane attack complex that causes direct lysis of the B cells by disrupting the cell membrane [3]. CDC is better mediated through the IgG1 constant region, and it has been demonstrated that CDC by rituximab correlates not only with the CD20 expression level but also with segregation of CD20 into specialized microdomains on the plasma membrane known as lipid rafts [7].
5.2.2.3 Apoptosis
Upon CD20 cross-linking after rituximab treatment, induction of apoptosis involving activation of caspase-9, caspase-3, and poly-adenosine diphosphate ribose polymerase (PARP) cleavage has been described in primary CLL cells, and downregulation of the antiapoptotic proteins XIAP and Mcl-1 has been described as well [8]. In addition, downregulation of the antiapoptotic protein bcl-2 by rituximab treatment has been observed in some B-cell lines [9]. Furthermore, it has been suggested that the transactivation of src family tyrosine kinases and subsequent increase of the intracellular calcium level upon CD20 translocation into lipid rafts are another proposed mechanism of action of rituximab-induced apoptosis [10, 11]. Additive and synergistic effects between rituximab- and chemotherapy-induced apoptosis have been observed suggesting that rituximab-induced apoptosis uses a final pathway similar to chemotherapeutic agents, leading to the use of combined chemoimmunotherapy regimens.
5.2.3 Susceptibility and Resistance to Rituximab
5.2.3.1 CD20 Expression
It has been observed that the CD20 expression level correlates with rituximab-mediated CDC. There are some in vitro data showing that CDC is more rapidly and efficiently induced by rituximab in cells with a higher CD20 expression level [12]. Therefore, loss or down modulation of CD20 may impact the efficacy of rituximab. As mentioned previously, CD20 is not shed or internalized following the binding of rituximab in normal individuals. However, it has been demonstrated that shaving/trogocytosis of CD20 is mediated by FcγR on acceptor cells, such as NK cells, monocytes, or macrophages in patients with CLL [13]. It has also been demonstrated that type I (rituximab-like) mAb-induced antigenic modulation occurs as a result of internalization of type I (rituximab-like) mAb-CD20 complexes by the B cells themselves in several cell lines [14] and the internalization is augmented by FcγRIIb [15]. In addition, CLL and mantle cell lymphoma (MCL) samples demonstrate more rapid internalization than follicular lymphoma (FL) and diffuse large B-cell lymphoma (DLBCL) [16], suggesting that internalization may explain the differing sensitivity of B-cell malignancies to rituximab.
5.2.3.2 Circulating Soluble CD20 (sCD20)
High levels of sCD20 are present in the plasma of patients with CLL. Plasma sCD20 has been shown to compete with cell surface CD20 for rituximab binding and significantly diminish the binding of rituximab to the surface of CLL cells [16]. It has been reported that response to rituximab is correlated with dose in patients with CLL, suggesting that rituximab dosing may be adjusted according to sCD20 level [17].
5.2.3.3 FcγR Polymorphisms
The influence of FcγR polymorphisms on rituximab-mediated ADCC has been previously described. There are three classes of FcγR: FcγRI (CD64), FcγRII (CD32), and FcγRIII (CD16). The polymorphism in FcγRIIIa consisting of a valine (V) or a phenylalanine (F) in amino acid position 158 results in a different affinity for IgG1-FcγRIIIa binding. The FcγRIIIa-158V homozygous genotype (V/V) has a greater affinity for human IgG1 than V/F or F/F. Patients with follicular lymphoma with the V/V genotype have a better clinical response to rituximab than patients with V/F or F/F. The polymorphism in FcγRIIa consisting of a histidine (H) or an arginine (R) in amino acid position 131 also results in a different affinity for IgG1-FcγRIIa binding, with better clinical results for FcγRIIa-131H homozygous patients. However, not all results demonstrate that these polymorphisms are clinically relevant [18–20]. It has been described that FcγRIIIa and FcγRIIa polymorphisms do not predict the clinical response to rituximab in patients with CLL [21].
5.2.3.4 Complement Regulatory Proteins
The role of complement regulatory proteins such as CD46, CD55, and CD59 in rituximab-mediated CDC has been previously described. Expression of high levels of CD46 or CD55 prevents the activity of C3 convertase, whereas expression of CD59 prevents the assembly of the active membrane attack complex, resulting in interference of completion of the complement cascade. There are some in vitro data showing that complement regulatory proteins may play an important role in rituximab-mediated CDC [22–24]. Currently, however, there is little in vivo data showing that CD46, CD55, or CD59 expression level correlates with response to rituximab.
5.2.4 Pharmacokinetics of Rituximab
Table 5.1 shows pharmacokinetic parameters, and Fig. 5.2 shows a serum concentration over time plot in patients with B-cell non-Hodgkin lymphoma, who received four or eight weekly doses of intravenous rituximab 375 mg/m2. From the first to the fourth or eighth intravenous infusion of rituximab, post-infusion mean serum concentrations increased and almost doubled. Steady-state serum concentrations of rituximab were not reached following four or eight weekly infusions. In addition, patients still had detectable rituximab concentrations in their serum several months after the end of treatment. Serum levels of free rituximab were measured by enzyme-linked immunosorbent assay (ELISA) [26].
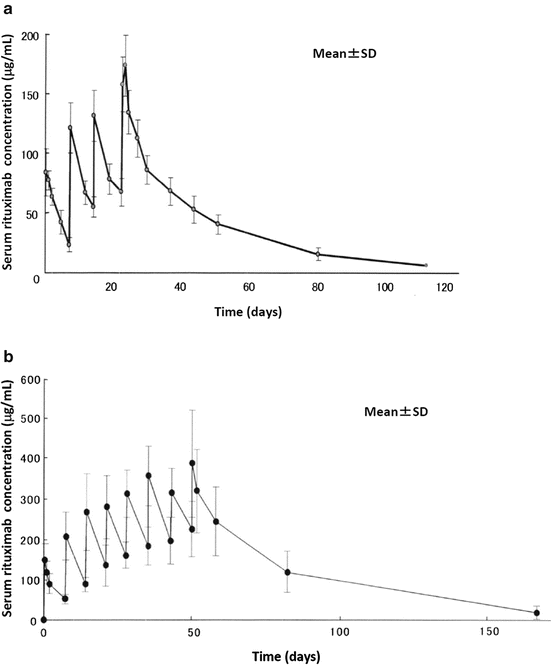
Table 5.1
Pharmacokinetic parameters of rituximab
Number of weekly doses of 375 mg/m2 | n | – | AUC (μg・h/ml) | C max (μg/ml) | t 1/2 (h) | CL (L/h) | MRT (h) | Vd (L) |
|---|---|---|---|---|---|---|---|---|
Four | 8 | Mean | 118,237 | 194.3 | 387.8 | 0.0437 | 517 | 11.16 |
SD | 49,963 | 54.5 | 176.6 | 0.0601 | 232 | 3.00 | ||
Eight | 15 | Mean | 502,147 | 445.2 | 393.6 | 0.0122 | 568 | 6.13 |
SD | 174,273 | 103.0 | 185.2 | 0.0038 | 267 | 1.55 |
Fig. 5.2
Pharmacokinetics of rituximab. Mean serum rituximab concentrations as a function of time following four weekly infusions of 375 mg/m2 (a, n = 8) or eight weekly infusions of 375 mg/m2 (b, n = 15). From the first to the fourth or eighth intravenous infusion of rituximab, post-infusion mean serum concentrations increased with time, and steady-state serum concentrations were not reached following four or eight weekly infusions
5.2.4.1 Distribution
It has been demonstrated that rituximab is widely distributed in body organs. By gamma camera measurements, radioactivity was detected in the heart, liver, lungs, spleen, and kidneys of patients with relapsed NHL who received iodine-131-labeled rituximab. The ratios of tumor to whole-body radioactivity increased over time, whereas those of organ to whole body decreased, indicating evidence of specific antibody binding [27]. It has been reported that rituximab levels in cerebrospinal fluid were detected after infusions with a C max of 0.55 μg/ml compared with a C max of 400 μg/ml in peripheral blood [28].
5.2.4.2 Metabolism
Little is known about the mechanisms of rituximab metabolism. It is speculated that rituximab is degraded by a nonspecific catabolism of proteins in the liver and other organs.
5.2.4.3 Excretion
It has been demonstrated that radioactivity was excreted mainly through the kidneys in patients with relapsed NHL who received iodine-131-labeled rituximab. The percentage of radioactivity excreted in the urine was almost 50% at the time of the whole-body half-life [27].
5.2.5 Overcoming Rituximab Resistance
5.2.5.1 Rituximab in Combination with Other Agents
The combined use of rituximab and other agents with different mechanisms of action may be one of the optimal solutions for overcoming rituximab resistance.
Fludarabine
It has been demonstrated that there is synergistic activity between rituximab and fludarabine, a purine analog agent, against a follicular lymphoma cell line. Fludarabine has been shown to induce downregulation of complement regulatory proteins CD46 and CD55 without significantly altering CD20 expression, resulting in enhancing rituximab-mediated CDC [29]. Although rituximab and fludarabine have demonstrated just an additive cytotoxic effect in primary CLL cells, the combination therapy of rituximab and fludarabine has already given positive results in the treatment of patients with CLL.
Bendamustine
It has been demonstrated that there is synergistic activity between rituximab and bendamustine, an alkylating agent, against primary CLL cells in vitro. Bendamustine has been shown to synergistically induce the proapoptotic effects of rituximab that may depend on caspase-7 and caspase-8 activation, but not p53 or caspase-3 [30]. Practically, it has been reported that the combination therapy of rituximab and bendamustine is active in patients with previously untreated, relapsed, or refractory CLL [31, 32].
Lenalidomide
It has been demonstrated that there is synergistic activity between rituximab and lenalidomide, an immunomodulatory agent, against CLL and B-NHL cells in vitro. It has been shown that lenalidomide enhances NK cell-mediated ADCC of rituximab-treated primary CLL cells that have been treated previously with fludarabine plus cyclophosphamide and that this enhanced NK cell-mediated ADCC is associated with enhanced granzyme B and Fas ligand expression [33]. It has also been demonstrated that lenalidomide repairs defective T-cell immune synapse formation, a characteristic of CLL, and enhances NK cell-mediated ADCC of rituximab-treated CLL cells [34, 35]. However, it has been demonstrated that lenalidomide promotes internalization of CD20 in CLL cells, resulting in decreased expression of cell surface CD20 [36]. These studies suggest that lenalidomide may act synergistically to enhance rituximab-mediated anti-CLL activity if alternative sequences of administration are optimized. Practically, it has been reported that the combination therapy of rituximab and lenalidomide is active in patients with relapsed or refractory CLL [37].
Histone Deacetylase (HDAC) Inhibitors
It has been demonstrated that HDAC inhibitors augment the cytotoxic activity of rituximab by increasing the surface expression of CD20 on lymphoma cells. Valproic acid and romidepsin have been shown to increase CD20 expression at protein and mRNA levels in B-cell lymphoma cell lines with relatively low expression of cell surface CD20, resulting in enhanced rituximab-mediated CDC [38, 39]. Entinostat has also been shown to increase CD20 expression at protein and mRNA levels in rituximab-sensitive or rituximab-resistant B-cell lymphoma cell lines and enhance rituximab activity in vivo [40]. However, these findings have not yet been extended to clinical trials in CLL.
5.2.5.2 Treatment with Other Anti-CD20 Monoclonal Antibodies
The use of other anti-CD20 mAbs, such as ofatumumab, obinutuzumab, tositumomab, 90Y-ibritumomab tiuxetan, and 131I-tositumomab, is another optimal solution for overcoming rituximab resistance.
5.2.6 Characteristic Toxicities and Management
5.2.6.1 Infusion Reactions
Infusion reactions are one of the most commonly occurring adverse reactions with rituximab. Infusion reactions are mediated by cytokine release (cytokine release syndrome), not by immunoglobulin E (IgE) (type 1 hypersensitivities). When cytokines, such as tumor necrosis factor-α (TNF-α), interferon-γ (IFN-γ), or interleukin-6 (IL-6), are released into the circulation from cells targeted by rituximab as well as immune effector cells recruited to the area, systemic symptoms such as rigor, fever, headache, nausea, sweating, itching, skin rash, cough, fatigue, hypertension, hypotension, tachycardia, angioedema, facial edema, hypoxemia, bronchospasm, pulmonary infiltrates, interstitial pneumonia, acute respiratory distress syndrome, myocardial infarction, ventricular fibrillation, and myocardial shock can occur. Infusion reactions usually occur within 30–120 min, most often with the first infusion. The symptoms are generally mild to moderate in severity and manageable in most patients. A slow initial rate of infusion is recommended to reduce the risk of infusion reactions. Prophylactic administrations of histamine antagonists (diphenhydramine) and antipyretic analgesics (acetaminophen) are also recommended. Steroids (hydrocortisone) should be considered in patients receiving rituximab not already in combination with steroids [41, 42].
5.2.6.2 Late-Onset Neutropenia (LON)
LON is a recognized late complication of rituximab-combined chemotherapy, occurring at least 4 weeks after treatment. The incidence of LON has been reported at about 25%, and a high incidence of LON has been reported in patients treated with an intensive primary chemotherapy regimen, such as high-dose therapy followed by stem cell transplantation [43]. LON has been attributed to rituximab, but the mechanism for developing LON remains unclear. It has been suggested that perturbation of stromal-derived factor-1 (SDF-1) during B-cell recovery following rituximab treatment retards neutrophil emergence from the bone marrow. SDF-1 is a chemokine required for early B-cell development and retention of the B-lineage and granulocytic precursors in the bone marrow microenvironment [44]. It has been demonstrated that the FcγRIIIa 158 V/V and V/F polymorphisms are associated with developing LON. This suggests that in patients with a high-affinity genotype, depletion of B cells may be more profound, subsequently stimulating increased bone marrow lymphopoiesis and resulting in less effective granulopoiesis [45]. The clinical course of LON was generally self-limited and not associated with severe infections.
5.2.6.3 Hepatitis B Virus Reactivation
Reactivation of hepatitis B virus (HBV) infection is a well-recognized complication in hepatitis B surface antigen (HBsAg)-positive cancer patients when they undergo cytotoxic chemotherapy. However, HBV reactivation has been reported to occur not only in HBsAg-positive patients but also in patients with a resolved HBV infection who are negative for HBsAg but positive for antibodies against the hepatitis B core antigen (anti-HBc) and/or antibodies against the hepatitis B surface antigen (anti-HBs) undergoing cytotoxic chemotherapy. HBV reactivation has been previously reported to be much less common in patients with resolved HBV, but with the recent increase in the use of rituximab, reports have increased. Since HBV reactivation in patients with resolved HBV is associated with a higher risk of fulminant hepatic failure, patients with resolved HBV receiving rituximab should be closely monitored with HBV DNA for a more prolonged period of time, even after completion of rituximab, so as to promptly start administration of nucleoside/nucleotide analogs to prevent HBV reactivation [46–50].
5.2.6.4 Opportunistic Infections
It has been observed that opportunistic infections, such as pneumocystis jiroveci pneumonia (PCP) or some viral infections, increase in patients receiving rituximab. However, the exact influence of rituximab on incidence of infection is still controversial. It has been suggested that, although rituximab by itself does not increase the incidence or change the spectrum of infections in hematologic patients, a possible influence of concomitant immunosuppressive medication such as chemotherapy and/or significant doses of glucocorticoids on the frequency of infections may be present. Screening and/or prophylaxis should be considered in patients receiving rituximab in combination with other immunosuppressive medications to minimize infectious complications. Sulfamethoxazole-trimethoprim prophylaxis against PCP is generally recommended [51].
5.3 Alemtuzumab
5.3.1 Development of Alemtuzumab
5.3.1.1 Background and Features
Alemtuzumab (Campath-1H) is a recombinant anti-CD52 humanized IgG1 kappa mAb (Fig. 5.3) [52]. A murine immunoglobulin M (IgM) antibody (Campath-1M) was initially selected, followed by a murine IgG2b antibody (Campath-1G) that demonstrated clinical activity in refractory CLL, even in patients who had failed Campath-1M [53]. Subsequently, a reshaped human IgG1 antibody, Campath-1H, was constructed by genetic engineering using the hypervariable regions from Campath-1G and found to have equivalent or superior activity to Campath-1G in vivo [54]. Alemtuzumab is the first mAb to be approved for use in the treatment of CLL.
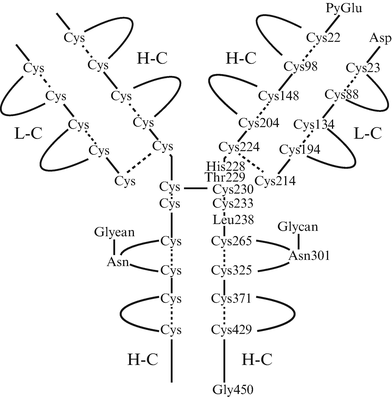
Fig. 5.3
Structure of alemtuzumab. Alemtuzumab is a recombinant anti-CD52 humanized IgG1 kappa monoclonal antibody constructed by genetic engineering using the hypervariable regions from murine antihuman CD52 IgG2a antibodies. The molecular formula is C6468 H10066 N1732 O2005 S40, and the molecular weight is approximately 150,000 daltons. H-C heavy chains, L-C light chains
5.3.1.2 CD52 Antigen
The CD52 antigen is a 21- to 28-kDa heavily glycosylated membrane-anchored glycoprotein with a short sequence of only 12 amino acids. Although the physiological functions of CD52 have yet to be clarified, it is thought to mediate a variety of biological effects such as the promotion of cell-cell adhesion, protection of the cell from environmental insult, activation of T-regulatory cells, and signal transduction. CD52 is highly expressed on all B and T lymphocytes at various stages of differentiation, except plasma cells, as well as on granulocytes, monocytes, macrophages, eosinophils, NK cells, and dendritic cells. The only other site of expression is in the male reproductive tract [55–57].
5.3.1.3 Favorable Features of CD52 as a Target Antigen
The CD52 antigen seems to be highly expressed on lymphocyte tumor cells, particularly in T-cell prolymphocytic leukemia (T-PLL), followed by CLL, with lower levels of expression on normal lymphocytes [58]. CD52 is not expressed on hematopoietic stem cells, plasma cells, erythrocytes, or platelets. Therefore, they are spared from the direct antibody effects [55–57].
5.3.2 Mechanism of Action of Alemtuzumab
Although the mechanism of action of alemtuzumab has yet to be clarified, proposed mechanisms of action of alemtuzumab include ADCC, CDC, and induction of apoptosis. ADCC is mediated by immune effector cells binding to alemtuzumab-coated cells, and this requires alemtuzumab to bind through the Fc region to FcγRs on immune effector cells. CDC is mediated by the recruitment of components of the complement cascade through the Fc region. Both ADCC and CDC are better mediated through the IgG1 constant region [55–57]. Alemtuzumab has been shown to induce not only caspase-dependent apoptosis but also caspase-independent and lipid raft-dependent apoptosis in primary CLL cells [59].
5.3.3 Susceptibility and Resistance to Alemtuzumab
5.3.3.1 CD52 Expression
It has been shown that patients who fail to respond to alemtuzumab have a lower CD52 expression, whereas the highest expression values are found in patients with a major response to alemtuzumab, suggesting that the level of CD52 expression correlates with alemtuzumab treatment [58].
5.3.3.2 Circulating Soluble CD52 (sCD52)
It has been demonstrated that high levels of sCD52 are present in the plasma of patients with CLL. Plasma sCD52 has been shown to be able to form immune complexes with alemtuzumab, resulting in reducing the serum concentration of alemtuzumab, and may influence the efficacy of alemtuzumab treatment. These findings suggest that patients with CLL and high sCD52 levels may require a higher dose of alemtuzumab [60].
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


