65 Retinoblastoma
Retinoblastoma was first described by Petras Pawius of Amsterdam as early as 1657. However, James Wardrop, an Edinburgh ophthalmologist, was officially credited for recognizing retinoblastoma in 1809, when he documented the first enucleation successfully performed to treat retinoblastoma.1 Two centuries later, enucleation is still used to treat retinoblastoma, along with other methods such as external beam radiation therapy, brachytherapy, cryotherapy, and laser therapy. Newer modalities that have recently gained popularity include chemoreduction and transpupillary thermotherapy. In many cases, treatment can help preserve both patients’ eyes and vision. In the United States, the current 5-year survival rate for children with retinoblastoma is 98%.2
Incidence
Surveys suggest a relatively constant occurrence of retinoblastoma during the last century throughout the world. The frequency of retinoblastoma has been documented as 1 in 14,000 to 1 in 34,000 live births.3 The incidence in the United States is relatively low, at 3.58 cases per million children under the age of 15 years, and is closely correlated with age. For ages 1 to 4 years, the incidence is 10.6 per million; for 5 to 9 years, 1.53 per million; and for 10 to 14 years, 0.27 per million.4 Approximately 350 new cases are diagnosed in the United States each year, and retinoblastoma is the seventh most common cancer in the pediatric population in this country.
Retinoblastoma appears to have no predilection for any particular racial group or gender. Retinoblastoma can affect one or both eyes. The right eye is affected as frequently as the left eye. Unilateral retinoblastoma occurs in 75% of all cases diagnosed, whereas bilateral disease occurs in 25% of cases. All patients with bilateral retinoblastoma have multifocal disease (more than one independent tumor focus in each eye) with 5 as the mean number of tumors distributed between the two eyes, and a range of 2 to 20 tumors. Of the patients with unilateral disease, only 15% have multifocal disease.5
Genetics
Retinoblastoma was one of the disease models that has been used to demonstrate the genetic nature of cancer. Knudson proposed the now classic two-hit model in 1971 after noting that the timing of tumor development (younger age at diagnosis of bilateral tumors than of unilateral tumors) suggested a mechanism in which at least two events would be responsible for the development of the tumor.6 He hypothesized that patients with multifocal bilateral disease were germline carriers for the first hit, with only one second hit necessary for the development of retinoblastoma. Unilateral unifocal patients, he determined, usually had a normal germline genome, but developed both hits in the progenitor tumor cell.
Since the Rb1 gene was isolated in 1986 by Friend et al. many studies have explored its location and function. The gene is located on the long arm of chromosome 13, band 14.2.7 Further characterization of the gene has revealed that it spans 200 kb and is composed of 27 exons. The gene encodes a 4.7 kb mRNA transcript, which is expressed in all adult tissues. The 110 kD nuclear phosphoprotein consists of 928 amino acids.
The protein encoded by the gene is a regulator at the cell cycle checkpoint between G1 and entry into the S-phase.8 The phosphorylation pattern of p110 RB varies during the cell cycle and the current model suggests that the unphosphorylated normal RB1 protein binds transcriptional regulators that promote entry into the S-phase. When the normal RB1 protein is phosphorylated, it dissociates from E2F, freeing it to bind to DNA and stimulate transcription of downstream genes that promote progression through the cell cycle. Loss of normal RB1 function, as in the case of the tumors, presumably allows for uncontrolled entry into the S-phase, more rapid cell cycling, and rapid cell division. The retinoblastoma protein is thus a dominant suppressor of tumor formation, making the Rb1 gene a member of the tumor suppressor class of genes.
Mutations in the Rb1 gene seem to occur throughout the gene. The majority of the germline mutations are nonsense or frameshift, introducing a premature stop codon and therefore producing a truncated and presumably nonfunctional protein. Other types of mutations such as missense, aberrant splice mutations, and mutations in promotor sequences, can result in a reduced penetrance or decreased expressivity form of the disease. Only about 2% to 3% of tumors demonstrate karyotypically visible larger deletions of material in the 13q14 band. Some of the children with this cytogenetic abnormality in their germline have a constitutional disease with severe developmental delay and dysmorphic features, named the 13q syndrome, whereas others are phenotypically normal other than their retinoblastoma. Evidence suggests that retinoblastoma, like other cancers such as colon carcinomas and glial brain tumors, requires other genetic abnormalities to occur before transformation into malignancy. This finding has stimulated many studies focused on subsequent mutations that occur after the initial mutation in the Rb1 gene. Candidate oncogenes and tumor suppressor genes include KIF14, MDM4, MYCN, E2F3, DEK, and CDH11.9
Genetic counseling for the families of retinoblastoma patients is complex and challenging (Table 65-1). Epidemiologic observation reveals that a parent with bilateral retinoblastoma has roughly a 45% chance of having each child with retinoblastoma, although the retinoblastoma may not be present at the child’s birth in either eye. Of those parents with bilateral disease whose children develop unilateral disease, 96% of the children will have multifocal tumors and most of them will be diagnosed within the first 6 months of life. Since all of these children (of bilateral parents) have a germinal mutation, 45% of their children will also develop retinoblastoma.
Table 65-1 Genetic Counseling for Retinoblastoma
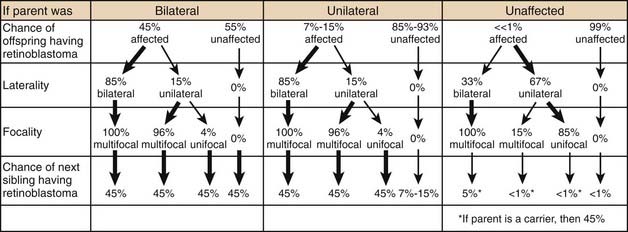
Two of the most common and challenging types of families who require genetic counseling are: (1) families in which neither parent has a history of retinoblastoma but the couple already has one child with the disease and wants to know the chances that the next will also have retinoblastoma, and (2) families in which one parent has a history of unilateral retinoblastoma and the couple wants to know the risk to future children of developing the disease. In the first scenario, an ophthalmoscopic examination of both parents’ eyes should be performed. The reason for this examination is that 1% to 2% of such parents will demonstrate on fundoscopic examination a rare benign clinical entity called a retinoma. There is debate as to the exact characterization of these lesions, but they likely represent either retinoblastoma precursor lesions as a result of the loss of the RB1 gene without the subsequent acquisition of other mutations that are required for progression to the malignant state or spontaneously regressed or arrested retinoblastoma.10 These parents should be counseled that although they have never been treated for retinoblastoma, they may harbor a germinal mutation, and if they have multifocal retinomas, each of their children will have a 45% chance of inheriting the mutation. Parents with no ophthalmoscopic evidence of retinoblastoma can be told that the chance that their next child will have retinoblastoma is between 1% and 5%. In the small percentage of cases in which the second child does develop the disease, it is presumed that one of the parents carries a germinal mutation although he or she is phenotypically normal. Several have been theories fashioned to explain these rare cases, including epigenetic modulation and somatic mosaicism in the parent. Additionally, some large kindreds have demonstrated the presence of two independent, distinct somatic mutations in different family members.
In these scenarios as well as others, parents of children with retinoblastoma are often eager to undergo genetic analysis to determine the risk that siblings or future children will develop the disease. Some centers have devised economical ways to routinely offer genetic screening to all retinoblastoma patients with the ability to detect sequence alterations in the Rb1 gene in about 80% to 90% of patients with a germinal mutation in peripheral blood or tumor tissue available.11 These programs utilize multiplex ligation–dependent probe amplification (MLPA) to detect large deletions or duplications, microsatellite analysis to detect loss of heterozygosity (LOH), and denaturing high-performance liquid chromatography (D-HPLC) analysis to detect point mutations and small insertions or deletions, and quantitative multiplex polymerase chain reaction (PCR) of short fluorescent fragments (QMPSF).
Finally, because the gene is expressed in all adult tissues and the RB1 protein plays such a vital role in controlling cell proliferation, all patients who carry a germinal Rb1 mutation are at risk for the development of additional nonocular cancers. These cancers can occur as late as 40 years or more after the initial development of retinoblastoma. The incidence of these cancers is significantly increased in patients who receive external beam radiation, especially when given before the age of 1 year. Patients should be educated about their risk factors for developing these cancers, as it is these cancers, not retinoblastoma, that are the leading cause of death in patients with the germinal RB1 mutation.12 See “Select Results” later in this chapter for more information on additional nonocular cancers.
Anatomy
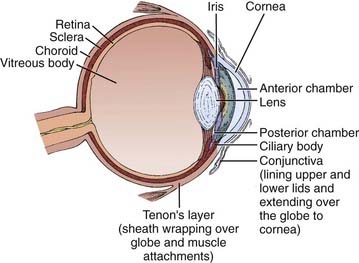
Figure 65-1 illustrates the important anatomic structures of the eye. The inner layer is the retina, where the photoreceptors are located. Retinoblastoma is thought to originate from an unidentified progenitor cell in this layer. The normal retina extends from the posterior “pole” forward to a region just behind the lens, in cross-section called the ora serrata. The middle layer of the eye is the choroid; this pigmented structure is continuous anteriorly with the ciliary body and iris, and together these are considered to be the uveal tract. The outermost layer of the eye is the sclera, a tough and radioresistant structure that fuses with the cornea anteriorly. The anterior chamber of the eye is between the cornea and the iris; the posterior chamber is just behind it, between the iris and the lens. Both chambers are filled with aqueous humor, produced by the ciliary body. Behind the lens is the vitreous chamber, with vitreous humor, a thick clear gel-like substance. In advanced retinoblastoma, “seeding” of the tumor is noted in this chamber.
Pathology
The diagnosis of retinoblastoma is routinely made without pathologic confirmation. Needle biopsies are rarely, if ever, indicated, as puncturing the eye can lead to extraocular extension and orbital invasion. Aqueous taps, previously explored as a possible diagnostic tool, are inappropriate for the same reason.13
When enucleation is the treatment of choice, histopathologic examination of the globe can be useful in providing information about potential extraocular extension (Fig. 65-2). For optimal prognostic information, the globe should be fixed for at least 24 hours, with the optic nerve preserved for separate evaluation.14
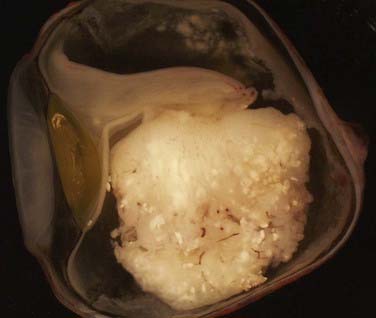
FIGURE 65-2 • Gross section of a globe, showing extensive disease in the retina, vitreous body, choroid, and optic nerve.
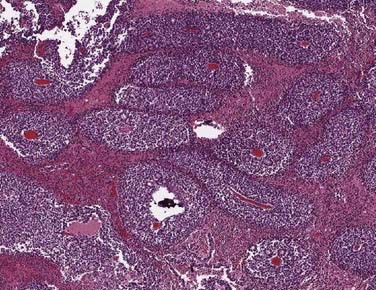
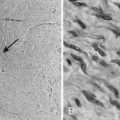
Under the microscope, the appearance of the retinoblastoma cells can range from small, round, undifferentiated cells with frequent mitoses, as seen in Fig. 65-3, to differentiated cells that form rosettes called Flexner-Wintersteiner rosettes.14 Important observations in assessing the risk of the child for distant or local relapse include careful assessment of the degree of invasion of tumor cells beyond the retina and into the choroid and sclera, and assessment of the degree of tumor invasion into the optic nerve beyond the region of the lamina cribrosa.15
Clinical Presentation
The presenting signs and symptoms of retinoblastoma vary depending on the geographic location in which the child presents. In developing countries, children can develop extraocular disease by the time they are diagnosed, with proptosis and an orbital mass. In these cases, the retinoblastoma has often extended directly into the orbit, causing rupture of the globe (Fig. 65-4). Regional nodal metastasis may be found in the preauricular or submandibular regions or the children may have widespread metastatic disease. These children are older at diagnosis (age 4 to 6 years) than patients in the United States, and few survive. In the United States, when there is no family history of disease, the median age at presentation is 1 year for bilateral retinoblastoma and 2 years for unilateral disease. Most children in the United States present with only intraocular disease and are diagnosed with signs rather than symptoms.
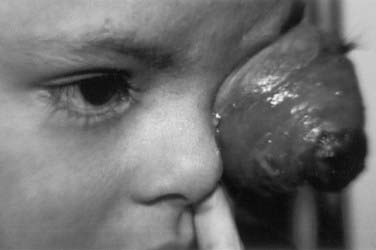
FIGURE 65-4 • A child with extensive orbital disease: the most common presentation in the developing world.
In the United States, the most common presenting sign of retinoblastoma (60% of cases) is leukocoria or a white pupillary reflex that is sometimes referred to as a cat’s eye reflex. Leukocoria can be the result of the tumor itself, a secondary retinal detachment associated with the tumor, or a reflection of light from the white mass at the posterior portion of the eye (Fig. 65-5).
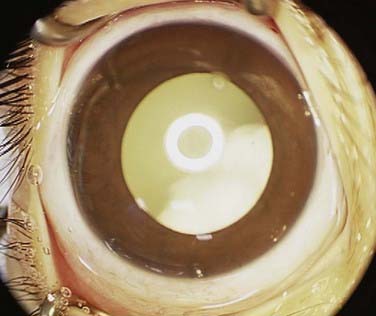
FIGURE 65-5 • A child with the “cat’s eye” reflex, with retinoblastoma visualized as the white material in the pupil.
The second most common sign is strabismus, misalignment of the two eyes. Of the 20% to 25% of patients who present with this sign, 10.5% have eyes crossed in (esotropia) and 5.2% out (exotropia).16 Esotropia in children is generally more common than exotropia, such that an infant with exotropia must be suspected to have retinoblastoma until proved otherwise. The strabismus in retinoblastoma patients is caused by tumor or retinal detachment located in the fovea of the eye, the area of central vision. Although patient survival is independent of whether patients present with strabismus or leukocoria, ocular survival rates are significantly lower for patients who present with leukocoria, as the disease tends to be more advanced at presentation in these cases.17
Presenting signs in the United States that occur in less than 5% of cases include: anisocoria (different-sized pupils); heterochromia (different-colored irides); hyphema (blood in the anterior chamber); tumor hypopyon (tumor in the anterior chamber); nystagmus, a unilateral fixed and dilated pupil; and failure to thrive. In cases in which the patient has gross chromosomal abnormalities, children can present with other findings, such as absence or hyperplasia of the thumbs, mental retardation, prominent nasofrontal bones, large malformed ears, hypospadias, bifid scrotum, and extra digits. Only 7% of patients are detected on routine pediatric screening by a pediatrician, whereas 72% of cases are detected by a family member or friend.17
When there is a family history of retinoblastoma, diagnosis tends to occur earlier, with a median age at diagnosis of 5 months for patients with unilateral disease and 11 months for patients with bilateral disease.18 Sixty percent of all retinoblastoma patients diagnosed under 6 months have no signs or symptoms, but are examined and diagnosed because of a family history of retinoblastoma.19 At our center, children with a family history of retinoblastoma typically undergo frequent serial fundoscopic examinations beginning within 24 to 48 hours of birth and continuing until at least 28 months of age. Twenty-eight months is chosen as the earliest possible time to stop examining these patients because it is the oldest age at which a patient with a positive family history of retinoblastoma has developed his or her first tumor in a previously documented disease-free eye.20 These serial examinations have a significant impact on ocular outcome. Patients who began undergoing screening examinations at our center as newborns because of a family history had a 68% ocular 5-year survival rate, whereas nonscreened patients with a family history had only a 38% ocular survival.17
The anatomic location of these tumors within the retina and the age at which the lesions develop have been well studied (Fig. 65-6). Retinoblastoma may be present anywhere in the retina at birth, but as age at tumor development increases, tumors develop progressively farther into the periphery. Thus macular tumors present earliest and peripheral anterior tumors present later. In our center, the average macular tumor is diagnosed at 5.6 months and never presents after 15.5 months. In contrast, peripheral tumors are diagnosed at an average of 16.4 months and can present as late as 8 years of age. This pattern of ocular tumor development has important implications for the radiation oncologist. Since many young children will have no tumors in the anterior retina, there is no need to treat this area, thereby decreasing the incidence of radiation-induced cataracts. However, future development of new peripheral tumors should be expected and the family should be informed of this likelihood. The development of these tumors does not represent a failure of radiation, but rather the inevitable process of tumorigenesis in genetically altered cells. Furthermore, the family of a child with multifocal unilateral retinoblastoma can be assured that new tumors that develop in the fellow eye will rarely originate in the fovea, so the outlook for vision in the fellow eye is typically favorable.
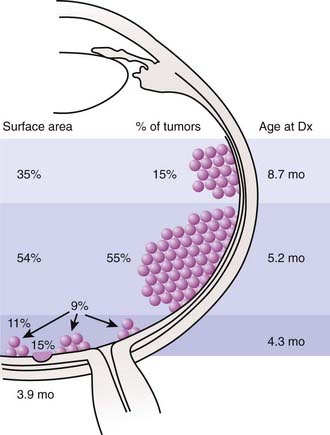
Routes of Spread
The natural course of untreated retinoblastoma follows one of five routes of progression: contiguous spread through the choroid, sclera, and orbit; extension along the optic nerve directly into the brain; invasion of the subarachnoid space and leptomeninges via the cerebrospinal fluid; hematogenous spread to bone marrow, bone, liver, and dissemination via conjunctiva through the lymphatic system.8 In the United States, the few patients with unilateral retinoblastoma who develop metastases usually have them at the time of diagnosis. The majority metastases occur within 5 years of diagnosis.18
Diagnosis of Retinoblastoma
All retinoblastoma tumors are drawn on an expanded concentric circle drawing of the globe called a fundus diagram, used to document the presence, location (in reference to the optic nerve and fovea of the eye), and size of the retinoblastoma tumors. Some ophthalmologists document the presence of tumors in the posterior pole by taking color photographs with a hand-held fundus camera while the patient is under anesthesia. Figure 65-7 demonstrates both techniques in the same child. The original tumor drawing is referred to during follow-up visits, and new drawings are composed to document treatment response. Although the ophthalmoscopic examination should always be considered the primary method for evaluation of retinoblastoma, several ancillary tests can assist in the diagnosis, especially when there is no clear view of the fundus.
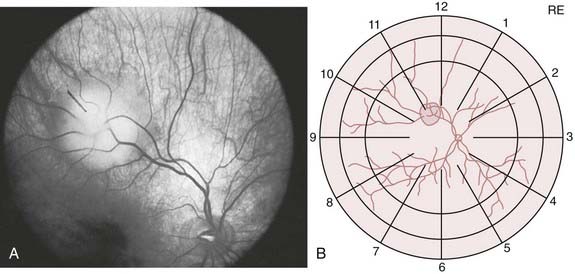
FIGURE 65-7 • A fundus photograph of an eye with retinoblastoma (A) and the corresponding sketch of the disease in the eye diagram (B).
Ophthalmic ultrasonography is one of the most frequently performed ancillary tests for retinoblastoma. It is noninvasive, safe, repeatable, and immediately interpretable. Ultrasonography should be performed on both eyes and can be performed with or without general anesthesia. B-scan ultrasonography reveals a two-dimensional (2D) cross-sectional view of the eye, confirms the presence and the relationship of the solid tumor to other anatomic structures within the eye, and detects the size and shape of the tumors (Fig. 65-8). B-scan can detect orbital involvement, optic nerve invasion (sometimes), extrascleral extension, and calcification. Because there is calcium (which exhibits high reflectivity on ultrasonographic scans) in the majority of retinoblastoma tumors, high amplitude echoes will remain on the screen even if the gain or sensitivity is lowered. Shadowing defects posterior to the tumor may be present and are caused by the absorption of sound and high reflectivity. A-scan ultrasonography evaluates the internal characteristics and vascularity of the tumor and measures the height of the tumor.
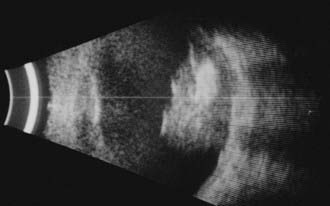
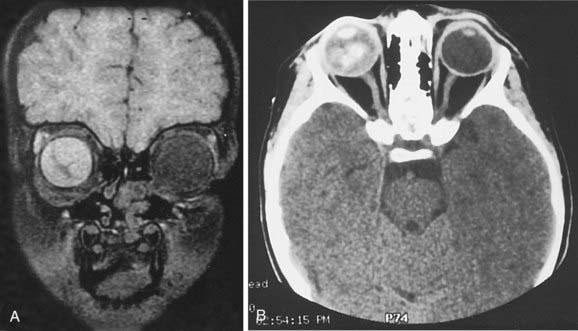
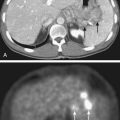
Computed tomography (CT) scans may no longer be routinely appropriate for retinoblastoma patients, as analysis has suggested an increased lifetime risk of other cancers in pediatric patients subjected to this imaging modality.21 Instead, as part of an extent-of-disease work-up, magnetic resonance imaging (MRI) is routinely performed (Fig. 65-9A, B). In addition to its excellent resolution in the diagnosis of extraocular soft tissue disease, MRI can readily distinguish between retinoblastoma and Coats’ disease (see “Differential Diagnosis of Retinoblastoma”), as Coats’ disease appears brighter than retinoblastoma on T2-weighted images due to proteinaceous exudate.8 One disadvantage of MRI is that calcification, a key feature of retinoblastoma, is more easily demonstrated with CT than with MRI.
Differential Diagnosis of Retinoblastoma
Retinoblastoma can be simulated by several other ophthalmic tumors and ophthalmic disorders (Table 65-2). The lesion that most commonly simulates retinoblastoma is Coats’ disease. Coats’ disease is a nonheritable, primarily unilateral anomaly that affects mostly boys, with a later age at diagnosis than retinoblastoma (mean age at diagnosis is 6 years). Ophthalmic examination reveals telangiectatic vessels of the retina with intraretinal and subretinal exudates and an exudative retinal detachment. Presence of a yellow-green sheen on indirect ophthalmoscopic examination aids in differentiating Coats’ from retinoblastoma. When rubeosis iridis, glaucoma, and corneal edema develop, the fundus may be impossible to view. Under these circumstances, ultrasonography can be helpful.
Table 65-2 Lesions Simulating Retinoblastoma
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


