The maintenance of gas exchange may be tenuous in the injured patient because of dysfunction in three key elements of the respiratory system. First, the central nervous system may be impaired, resulting in inadequate respiratory drive, or inability to maintain patent proximal airways. Second, injury to the torso can produce changes in compliance, ineffective respiratory effort, and pain that impact the patient’s ability to complete the work of breathing. Third, primary and secondary insults to the lung result in ineffective gas exchange. In practice, it is common for patients to suffer simultaneous insults, affecting all three elements. Respiratory failure that relates primarily to CNS injury is discussed at length in other chapters and will not be extensively covered here. This chapter will focus on insults that affect work of breathing and gas exchange. The syndrome of acute respiratory distress syndrome (ARDS) and current management is a major focus.
The neurohormonal response to injury (Chapter 61) results in a remarkable increase in cellular metabolism. This creates a substantial increase in carbon dioxide (CO2) production that must be matched by increased elimination from the lungs. While a resting adult eliminates 200 cm3/kg/min of CO2, post-injury hypermetabolism results in CO2 production in the range of 425 cm3/kg/min.1 Thus, the minute ventilation required to maintain eucapnia may rise from a resting rate of approximately 5 L/min to more than 10 L/min. This represents a 100% increase in the work of breathing simply to meet metabolic demands.
Additionally, injured patients typically have an increase in physiologic and anatomic dead space regions that do not participate in gas exchange. In a normal adult, the proportion of each breath that is dead space (Vd/Vt) is approximately 0.35. In the intubated, ventilated patient, the Vd/Vt can be accurately determined using Fowler’s nitrogen washout technique2 or more commonly calculated by a number of techniques including the Bohr–Enghoff method (Vd/Vt = [{Paco2 – mean expired CO2}/Paco2]).3 For practical purposes (since mean expired CO2 is not commonly measured), this is a reflection of the minute volume required to achieve a given Paco2. In ventilated patients with pulmonary failure, the Vd/Vt often exceeds 0.6. Simply put, this extra dead space is a burden because each breath is less effective at eliminating CO2, and therefore minute ventilation requirements in the 12–20 L/min range are not uncommon in the post-injury setting.
The above increase in respiratory demand might be met by a healthy adult; however, the injured patient faces several challenges in completing this additional work. CNS dysfunction from injury impairs respiratory drive, as do many medications routinely used for sedation and analgesia. Decreased thoracic compliance from abdominal distension (eg, as part of the abdominal compartment syndrome, Chapter 38), chest wall edema, and recumbent positioning increase the energy required to complete a respiratory cycle. Decreased pulmonary compliance from an increase in extravascular lung water and pleural collections (effusions/hemothorax) also contribute. Muscular weakness from impaired energetics (hypothermia, acidosis, coagulopathy, cardiovascular failure, mitochondrial dysfunction, and oxidant stress) or fatigue may be an insurmountable challenge. Finally, pain from torso injuries or operative interventions make the increased ventilatory demand a substantial burden to the patient.
The net effect of increased demand and diminished capacity to execute the work of breathing is hypercapnic respiratory failure. In the early post-injury period, patients most commonly present with a mixed acid-base disorder where ventilation is inadequate to maintain physiologic pH in the face of a metabolic acidosis. This frequently occurs in the absence of major hypoxia, and therefore caution is warranted in relying on oximetry alone to assess adequacy of pulmonary function. Particularly in patients with tachypnea, blood gas analysis is optimal to quickly identify patients who are not meeting their ventilatory demands. While efforts to diminish the work of breathing should be routine, most patients with hypercapnic failure require some form of mechanical ventilation to meet their demands. Approaches to invasive and noninvasive ventilation are covered extensively in Chapter 55.
Two injury patterns that precipitate hypercapnic respiratory failure are worthy of special mention: spinal cord injury and flail chest/pulmonary contusion. In spinal cord injury, conventional wisdom asserts that lesions below C5 should not result in pulmonary failure, because innervation to the diaphragm remains intact. In practice, however, most complete cord lesions in the cervical and upper thoracic regions routinely result in failure requiring mechanical ventilation.4 The genesis is multifactorial, including loss of vital capacity, ineffective cough with retention of secretions due to loss of innervation of intercostal and abdominal musculature, pneumonia in the setting of multiple injuries, and aspiration at the time of the initial insult. Furthermore, vagal nerve autonomic dysfunction results in increased secretions, bronchospasms, and pulmonary edema. This patient population requires aggressive mobilization and pulmonary care, as recurrent lobar collapse and pneumonia are certain and lethal if not aggressively prevented.
Early operative stabilization of the spine is advocated as it has been shown to decrease the need for mechanical ventilation and ICU stay.5 Other adjuncts such as noninvasive ventilation, bronchodilators, mucolytics, and percussion should be considered. Laparoscopic placement of diaphragmatic pacers is a benefit in select patients with a recent report by Onders and colleagues showing that 96% of SCI were able to use diaphragm pacing stimulation to provide ventilation replacing their mechanical ventilator.6 Largely, this has been studied in the rehabilitation setting, and therefore a role for this approach in the acute setting remains undefined.
Flail chest and pulmonary contusion can be thought of as a single entity. This is a challenging injury pattern, because it impacts both the patient’s ability to execute the work of breathing (from pain and mechanical instability of the thoracic) and gas exchange (from the pulmonary contusion). Isolated pulmonary contusion rarely requires mechanical ventilation. Since minor contusions are frequently identified by computed tomography (CT), it is important to realize that this tends to take a relatively benign course (see Chapter 25). It is clear that the number of rib fractures is strongly associated with the development of pulmonary failure, ARDS, and mortality, that profoundly impact the elderly population.7 Early pain control, preferably beginning in the emergency department with regional anesthesia, has been shown to be effective in reducing the impact of multiple rib fractures and should be routinely applied especially in those patients over the age of 65 that have more than three continuous ribs fractured.8 Admission to a high-volume trauma center, use of patient-controlled analgesia, epidural or paravertebral catheter administered analgesia, routine tracheostomy, and an algorithm-driven approach are associated with improved survival.9,10
Hypoxic pulmonary failure is a substantial cocontributor to respiratory failure in the trauma setting. The etiologies are diverse including aspiration pneumonitis, pneumonia, pulmonary embolism, congestive heart failure, blunt thoracic trauma, and ARDS. Most of these entities are discussed elsewhere; however, ARDS will be the focus of this chapter. Indeed, the mechanisms at work in ARDS share many common features with these other processes that affect the alveolar–capillary interface.
ARDS is a clinical syndrome of inflammatory lung injury that can be thought of as the final common pathway of diverse systemic processes. In the past two decades major progress has been achieved in defining the underlying ARDS pathophysiology and optimal supportive care. Specific therapies that address the underlying mechanisms responsible for ARDS development remain elusive at present.
The recognition of ARDS as a distinct clinical entity resulted from the description by Ashbaugh et al in 1967.11 Subsequent descriptions include five principal criteria: (1) hypoxemia refractory to oxygen administration; (2) diffuse, bilateral infiltrates on chest radiograph; (3) low static lung compliance; (4) absence of congestive heart failure; and (5) presence of an appropriate at-risk diagnosis.
Since the initial description of ARDS 50 years ago, the optimal definition of ARDS remains a controversial subject. The lung injury score (LIS), proposed by Murray and colleagues in 1988 has been a commonly used measure of lung injury severity in previous clinical studies. The LIS is based on four components: chest radiograph, hypoxemia, positive end-expiratory pressure (PEEP), and respiratory compliance (Table 57-1).12 Each component is scored from 0 to 4. The LIS is calculated by summing the scores of the available components and dividing by the number of components used. ARDS (or severe lung injury) is defined as an LIS greater than 2.5. Zero represents no lung injury and 0.1–2.5 represents mild to moderate lung injury.
Chest radiograph score No alveolar consolidation Alveolar consolidation confined to one quadrant Alveolar consolidation confined to two quadrants Alveolar consolidation confined to three quadrants Alveolar consolidation confined to four quadrants | 0 1 2 3 4 |
Hypoxemia score Pao2/Fio2 ≥300 Pao2/Fio2 225–299 Pao2/Fio2 175–224 Pao2/Fio2 100–174 Pao2/Fio2 <100 | 0 1 2 3 4 |
PEEP score (when ventilated) PEEP ≤5 cm H2O PEEP 6–8 cm H2O PEEP 9–11 cm H2O PEEP 12–14 cm H2O PEEP ≥15 cm H2O | 0 1 2 3 4 |
Respiratory system compliance score (when available) Compliance ≥80 mL/cm H2O Compliance 60–79 mL/cm H2O Compliance 40–59 mL/cm H2O Compliance 20–39 mL/cm H2O Compliance ≤19 mL/cm H2O | 0 1 2 3 4 |
The first standard definitions were developed in 1994, by the Consensus Conference of American and European investigators (AECC). Acute lung injury (ALI) was defined as respiratory failure of acute onset with a Pao2/Fio2 ratio less than or equal to 300 mm Hg (regardless of the level of positive end-expiratory pressure, PEEP), bilateral infiltrates on frontal chest radiograph, and a pulmonary capillary wedge pressure less than or equal to 18 mm Hg (if measured) or no evidence of left atrial hypertension. ARDS was defined identically except for a lower limiting value of less than 200 mm Hg for Pao2/ Fio2 (Table 57-2).13 The AECC definition of ALI/ARDS has been used in many of the ARDS Network clinical trials; however, debate still exists as to the usefulness of the AECC diagnostic criteria. Criticism of the AECC definition includes lack of account of the mode of ventilation and the level of PEEP, which can significantly affect oxygenation. Furthermore, with the decreased use of Swan–Ganz catheters, the wedge pressure is not commonly measured.14
Acute lung injury criteria Timing: acute onset Oxygenation: Pao2/Fio2 ≤300 mm Hg (regardless of positive end-expiratory pressure) Chest radiograph: bilateral infiltrates on anteroposterior chest radiograph Pulmonary artery occlusion pressure: ≤18 mm Hg or no clinical evidence of left atrial hypertension ARDS criteria Same as acute lung injury except: Oxygenation: Pao2/Fio2 ≤200 mm Hg (regardless of positive end-expiratory pressure) |
An alternative definition of ARDS developed by a consensus panel of senior investigators using the Delphi method appears to be more specific than AECC critiera.15 Briefly, the authors include PEEP in the consideration of hypoxia and require either the absence of CHF or the presence of a recognized risk factor for ARDS. The degree of hypoxia required is a P/F less than 200 with PEEP greater than or equal to 10; therefore, it excludes patients who are hypoxic purely because of de-recruitment or suboptimal PEEP (Table 57-3). By allowing patients with high filling pressures in the presence of a recognized ARDS risk factor, the definition recognizes the prevalence of high LAPs during the course of ARDS and includes patients with concomitant CHF and ARDS.
The definition of ARDS was most recently revised by a panel of specialists in a joint effort of the European Society of Intensive Care Medicine, the American Thoracic Society, and the Society of Critical Care Medicine. Their collective aim was to address the current limitations of the AECC definition and explore other defining ARDS variables. The panel agreed to maintain the previous conceptual model, which defined ARDS as a syndrome of acute, diffuse lung inflammation, with edema because of increased permeability of the alveolar–capillary membrane, and clinically characterized by decreased oxygenation because of increased venous admixture, decreased lung compliance, increased physiological dead space, and bilateral radiographic opacities. The newest and most widely accepted ARDS criteria, also referred to as the Berlin definition of ARDS, are as follows in Table 57-4.16 In a large, multi-ICU cohort of patients with ARDS, both LIS and the Berlin definition severity stages were associated with increased in-hospital morbidity and mortality; however, the predictive validity of both scores is only marginally better than chance alone.17
Timing: Within 1 week of a known clinical insult or new or worsening respiratory symptoms Chest imaging: Bilateral opacities—not fully explained by effusions, lobar/lung collapse, or nodules Origin of edema: Respiratory failure not fully explained by cardiac failure or fluid overload Oxygenation Mild: 200 mm Hg Moderate: 100 mm Hg Severe: Pao2/Fio2 <100 mm Hg with PEEP or CPAP ≥5 cm H2O |


The estimated incidence of ARDS in population-based studies is on the order of 40 cases per 100,000 person-years.18,19,20 In the United States, this represents about 200,000 cases per year, and about 15% of ICU admissions. With modern mortality rates (see further), one can estimate this entity is responsible for about 50,000 deaths per year in the United States.
Clinical risk factors for ARDS can be broadly categorized into direct and indirect pulmonary factors (Table 57-5). Direct pulmonary factors are those primarily associated with local pulmonary parenchymal injury and include inhalation injury, pulmonary contusion, aspiration, and pulmonary infection. Indirect pulmonary factors are those thought to be associated with systemic inflammation and resultant lung injury. These include trauma, shock, burn injury, extrapulmonary sepsis, transfusion of blood products, multiple long bone fractures, pancreatitis, and others.18,21
The King County Lung Injury Project (KCLIP) identified pulmonary and extrapulmonary sepsis as the most common risk factor with 46% and 33% of ARDS cases, respectively. Following sepsis in descending order of frequency were aspiration (11%), trauma (7%), transfusion (3%), drug overdose (3%), and pancreatitis (3%). Cases with another or no risk factor identified constituted 14% of the total. The highest mortality rates were noted in aspiration-associated (44%) and pulmonary-sepsis–associated lung injury groups (41%) with the lowest rate in trauma-associated lung injury (24%).20
Several demographic factors have been identified as risk modifiers for development of ARDS. The incidence of ARDS increases with age and older patients have consistently higher mortality rates, although it is unclear whether this simply represents diminished physiologic reserve in older patients.22,23 Alcohol abuse, active cigarette smoking, and exposure to second-hand smoke have also been linked to ARDS incidence.24,25 Higher BMIs have been associated with increased incidence of ARDS26 with data suggesting a decrease in mortality.27
It is well established that high tidal volume ventilation increases mortality for patients with established ARDS.28 Several studies have shown that increased tidal volume is an independent risk factor for the subsequent development of ARDS in a dose-responsive manner.29,30 This phenomenon, termed ventilator-induced lung injury (VILI), has been an area of increasing scrutiny.
Transfusion of blood products is a major iatrogenic risk factor for ARDS. Multiple studies have shown increased risk of ARDS in ICU patients receiving packed red blood cells, fresh frozen plasma, and/or platelet transfusions,31,32 a phenomenon, termed transfusion-related acute lung injury (TRALI). The National Heart, Lung, and Blood Institute established a consensus definition to promote research in TRALI. In addition to the standard criteria for ARDS, TRALI requires additional criteria (Table 57-6). A patient must (1) develop ARDS during or within 6 hours of transfusion and (2) no ARDS may be present before transfusion. (3) Alternative ARDS risk factors may be present, but the patients’ clinical course should determine whether the ARDS is mechanistically related to the transfusion.33
Onset of signs or symptoms ≤6 h after transfusion. No ALI may be present prior to transfusion. Alternative ALI risk factors may be present, but the patients’ clinical course should determine whether the ALI is mechanistically related to the transfusion. |
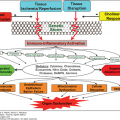
The current paradigm of systemic inflammation leading to ARDS posits that a variety of insults, both infectious and noninfectious, can result in an unbridled hyperinflammatory response. This leads to organ injury from indiscriminate activation of effector cells that subsequently release oxidants, proteinases, and other potentially autotoxic compounds. If the initial insult is severe enough, early organ dysfunction results (“one-hit” or single insult model). More often, a less severe insult results in a systemic inflammatory response that is not by itself injurious. These patients appear, however, to be primed such that they have an exaggerated response to a second insult, which leads to an augmented/amplified systemic inflammatory response and multiple organ dysfunction (“two-hit” or sequential insult model, see Chapter 61).34
Inflammatory models provide a unifying hypothesis for ARDS and MOF; however, the precise relationship between ARDS and MOF remains to be further refined. MOF is a frequent occurrence and the most common cause of mortality in patients with ARDS.35 Indeed, post-injury ARDS appears to be an obligate precursor of other organ failures.36 This may be because the lung is a primary target of the inflammatory process, or because the resultant pulmonary damage impairs the lung’s ability to metabolize inflammatory mediators and control cellular effectors of injury.37 It is also now clear that ventilator strategies that inadvertently promote lung injury may produce systemic inflammation, perhaps leading to other organ failures.38
Inflammatory lung injury leads to the pathologic lesion of diffuse alveolar damage. This prototypic lesion of ARDS is at the alveolocapillary interface, which results in epithelial and endothelial damage as well as high-permeability pulmonary edema. The histologic appearance of this lesion can be divided into three overlapping phases: (1) the exudative phase, with edema and hemorrhage; (2) the proliferative phase, with organization and repair; and (3) the fibrotic phase.39
The exudative phase generally encompasses the first 3–5 days but may last up to a week. The initial histologic changes include interstitial edema, proteinaceous alveolar edema, and intraalveolar hemorrhage. The exudative phase is characterized by the appearance of hyaline membranes, which are composed of fibrin, immunoglobulin, and complement. Electron microscopy reveals endothelial injury with cell swelling, widening intercellular junctions, and increased pinocytotic vesicles. In addition, there is disruption of the basement membrane.
The alveolar epithelium usually exhibits extensive loss of type I cells, which slough and leave a denuded basement membrane. While some loss may be from necrosis, it appears that apoptosis contributes substantially. Activation of matrix metalloproteinases, toll-like receptors, and oxidative stress pathways initiate programmed cell death in these cells. Demonstration of soluble Fas ligand in bronchoalveolar lavage (BAL) fluid early in ARDS supports this concept.40 Loss of the alveolar epithelial barrier results in alveolar edema, as the remaining cells are unable to drive sodium from the alveolar into the interstitial compartment.
During the proliferative phase, typically occupying the second 2 weeks after the onset of respiratory failure, type II cells divide and cover the denuded basement membrane along the alveolar wall. This process may be seen as early as 3 days after the onset of clinical ARDS. Type II cells are also capable of differentiating into type I epithelial cells. Fibroblasts and myofibroblasts proliferate and migrate into the alveolar space in the third phase. Fibroblasts change the alveolar exudate into granulation tissue, which subsequently organizes and forms dense fibrous tissue. Eventually, epithelial cells cover the granulation tissue. This whole process is called fibrosis by accretion and is important in lung remodeling. Septal collagen deposition by fibroblasts and “collapse induration” also contribute to fibrous remodeling of the lung in ARDS.
Many patients with ARDS recover lung function 3–4 weeks after initial injury; however, some enter a fibrotic phase characterized by thickened, collagenous connective tissue in the alveolar septa and walls. Pulmonary vascular changes occur as well, with intimal thickening and medial hypertrophy of the pulmonary arterioles. Complete obliteration of portions of the pulmonary vascular bed is the end result. Clinical sequelas include an increased risk of pneumothorax, decreased lung compliance, and increased pulmonary dead space. This may lead increased long-term support on mechanical ventilators and/or need for supplemental oxygen.
Lung injury in ARDS involves components of inflammation, coagulation, vasomotor tone, and other systems (see Chapter 55). The pivotal cellular mediators appear to be leukocytes, with both local and humoral mediators orchestrating their function. Activation of these leukocytes results in release or activation of multiple cytokines, chemokines, oxidants, and proteases that result in the final common pathway of tissue injury in ARDS.
A consistent histopathologic feature of ARDS is neutrophil infiltration of the pulmonary microvasculature, interstitium, and alveoli. Neutrophils infiltration may result in lung injury due to neutrophil-mediated release of reactive oxidant species (ROS) and resulting oxidant stress. Furthermore, neutrophils are an important source of proinflammatory cytokines. Persistence of neutrophils in serial BAL fluid samples from patients with ARDS suggests unbridled inflammation and portends poor prognosis. In animal models, neutrophil depletion prior to an insult markedly attenuates resulting lung injury.41 Evidence from clinical studies supporting excess ROS in ARDS include findings of increased hydrogen peroxide in exhaled breath,42 decreased levels of glutathione in lung lavage fluids,43 and increased lipid peroxidation products in plasma.44
The lung contains large numbers of fixed tissue macrophages that are a critical component of the inflammatory response in ALI. Activated macrophages can cause tissue injury by releasing the same toxic mediators as neutrophils (ROS and proteases). Probably more important is the macrophage capability to synthesize multiple proinflammatory mediators, such as complement fragments, cytokines, and chemokines. Thus, macrophages are thought to have a major role in amplifying and perpetuating the inflammatory response. This is exacerbated by the long half-life of the macrophage, which is measured in days rather than hours as in the neutrophil. The alveolar macrophage has two additional key functions: control of local infection and modulation of fibrosis.45 Alveolar macrophage from ARDS patients demonstrate defective phagocytosis and bacterial killing, reflecting an increased risk for infection in these patients.
The pulmonary endothelium is not a passive bystander in the pathogenesis of ARDS, but actively participates in initiating and perpetuating the inflammatory response. Endothelial cells increase the expression of adhesion molecules (ICAM-1, ICAM-3, and E-selectin) following exposure to an activating stimulus. These ligands serve as tethering and signaling molecules by binding to their cognate leukocyte membrane proteins. Thus, the endothelial cell actively coordinates trafficking, firm adhesion, and transmigration. In the setting of systemic inflammation, inappropriate endothelial cell activation may lead to indiscriminate leukocyte recruitment and parenchymal inflammation. Moreover, endothelial cells produce and release vasoactive substances, such as prostacyclin, nitric oxide (NO), and endothelins. These substances may mediate much of the pulmonary vascular dysfunction characteristic of ARDS. The activated endothelium expresses procoagulant activity, which also contributes to intravascular coagulation and microvascular dysfunction.46 Thrombin, in turn, has proinflammatory effects on leukocytes. Endothelial injury, then, may be both a proximate cause and a marker for ALI.
It has long been understood that reperfusion of the ischemic gut can lead to lung injury. Deitch et al have shown that injured intestine releases danger signals, transported via lymph, that induce inflammatory pathways resulting in organ injury.47 As the lung is the first organ exposed to mesenteric lymph (via the thoracic duct), it might explain the clinical observation that the lung is often the first organ to fail. In animal models, diversion of gut lymph appears to abrogate lung injury.48 While the precise mediators of this phenomenon are as yet unknown, changes in posthemorrhagic shock lymph flow, lipid content, and protein content are an active area of investigation.49
Systemic complement activation secondary to trauma or sepsis is considered a major early factor in ARDS.50 C5a, a product of complement activation, is a powerful neutrophil chemoattractant. Moreover, C5a induces neutrophil aggregation and activation leading to pulmonary neutrophil sequestration and lung injury. Clinically, plasma and bronchoalveolar C3a levels correlate with the development of ARDS.51
As mentioned above, the pulmonary endothelium is recognized as an active participant in the development of ALI. As such, markers of endothelial activation or injury have been investigated as predictors of the development of ARDS. von Willebrand factor antigen (vWF:Ag) has been studied fairly extensively as a marker of endothelial dysfunction. vWF:Ag is synthesized largely by vascular endothelial cells and has been shown to be a sensitive marker of endothelial injury or activation52 and predictive for the development of ALI.53
Following activation, endothelial expression of adhesion molecules, including ICAM-1, VCAM-1, E-selectin, and P-selectin, is upregulated. These compounds are susceptible to proteolytic cleavage and may exist in the circulation in a soluble form. Therefore, these molecules represent a measure of endothelial activation or damage. Elevated ICAM-1 levels have been found in severely injured patients who subsequently developed MOF.54 In contrast, plasma levels of soluble E- and P-selectins measured at admission were not useful in predicting lung injury.
Markers of leukocyte activation have also been measured in plasma and BAL fluid of patients in an attempt to predict development of ARDS. Gordon et al noted markedly elevated plasma elastase levels very early after multisystem trauma.55 Subsequent studies supported a causative role for neutrophil elastase in ARDS and have led to the clinical development of human elastase inhibitor (see the section “Pharmacologic Therapy”).56
Circulating and BAL fluid levels of cytokines are also inherently attractive as predictors of ARDS; whether they are markers or mediators remains an important question. Direct measurement BAL or pulmonary edema fluid in ARDS patients has shown that both fluids consistently contain elevated levels of TNF-α and IL-1; additionally, high levels of TNF-α and IL-1 in plasma and in BAL have been associated with an increased risk of death in patients with ALI and ARDS.57,58 Persistently elevated IL-6 levels have also been associated with an increased risk of death in ARDS patients. In the ARDSNet trial, there was a significantly steeper fall in IL-6 levels over the initial 72 hours in the lung protective treatment group with decreased mortality compared to the higher-tidal-volume treatment group.59 IL-10 is a potent inhibitor of proinflammatory molecules including TNF-α and IL-1. Lower levels of IL-10 in blood and lung lavage fluid have been in association with development of ARDS and associated with mortality.60
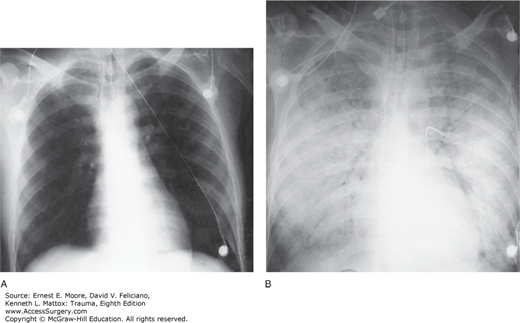
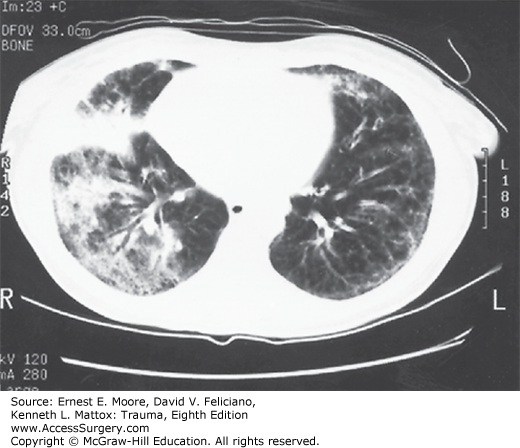
ARDS is characterized by diffuse, patchy, panlobular pulmonary infiltrates on plain chest radiograph (Fig. 57-1). CT of the chest will demonstrate that the parenchymal changes are inhomogeneous with the dependent lung regions most affected (Fig. 57-2). The inhomogeneous distribution of parenchymal densities led to the concept of a three-compartment model of the lung in ARDS.61 One compartment is substantially normal (healthy zone), one is fully diseased without any possibility of recruitment (diseased zone), and, finally, the third compartment is composed of collapsed alveoli potentially recruitable with increasing pressure (recruitable zone). Increased airway pressure is necessary to recruit collapsed alveoli. In the 1990s, however, it was already recognized that in the heterogeneously injured lung, VILI may be damaging to the healthy zone. This injury is thought to be responsible for severe protracted ARDS, as well as perpetuation of systemic inflammation and MOF.29,30
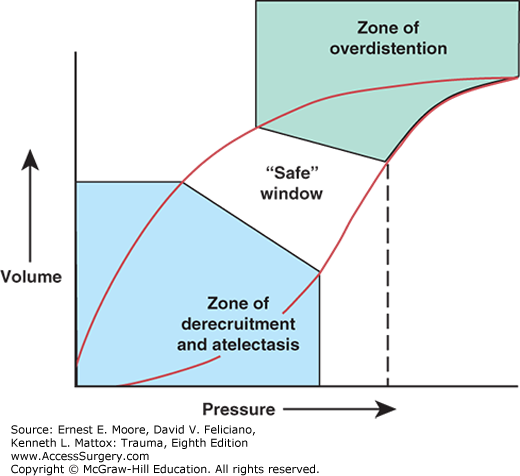
A standard method used to describe the mechanical properties of the lung is to determine the static pressure–volume (PV) curve during inflation and deflation (Fig. 57-3). In early ARDS, the lower inflection point represents the airway pressure at which considerable alveolar recruitment occurs. The upper inflection point is where near maximal inflation occurs such that further increases in airway pressure result in alveolar overdistension and little change in volume. In practice, however, inflection points in individual patients have been difficult to consistently measure.
FIGURE 57-3
Idealized static pressure–volume curve of the lungs in ARDS. At low pressures and volumes, derecruitment, atelectasis, and lung injury from repetitive opening and closing of gas exchange units are a concern. At high pressures and volumes, overdistention, increased shunt, and lung injury from excessive stretch predominate. In between these two extremes is the optimal zone where lung stays recruited but is not overstretched.
Related posts:



Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


