Fig. 1
The balance of DNA damage and DNA repair determines the stability of a genome
It is becoming more and more apparent that the fine tuning of genomic stability is not only a very important aspect of a species’ evolutionary potential but that changing the level of genomic stability in tumor cells has a great influence on the “evolutionary” potential of a tumor and thus on its ability to adapt to different external challenges like chemotherapy or to its ability to find appropriate niches for growth in the body (metastases). Recent in-depth sequencing studies and careful evaluation of traditional molecular genetic and cytogenetic data have revealed many examples of astounding clonal heterogeneity in tumors and provided evidence that tumor development is governed by the evolutionary principle of selection of the fittest (Heselmeyer-Haddad et al. 2012; Yates and Campbell 2012). It is therefore not surprising that we find many tumors with elevated levels of genomic instability, which allow the tumors to evolve more rapidly.
In this context, we need to take a close look at the factors that determine genomic stability and that are not only important for the generation of recurring chromosomal translocations but these factors can also be altered as a consequence of chromosomal translocations.
3 DNA Damage and Repair
3.1 Balance of DNA Damage and DNA Repair
The stability of a genome is determined by the balance of two factors (Fig. 1): (1) the rate of DNA damage and (2) the rate of DNA repair.
3.2 Sources of DNA Damage
There are many causes of DNA damage. Most commonly one thinks of DNA damaging external agents like ionizing radiation or ultra violet radiation, as well as chemicals or drugs like alkylating agents. However, spontaneous chemical reactions and cell internal sources of DNA damaging agents like reactive oxygen species (ROS) or S-adenosyl-methionine and errors in normal DNA metabolism (transcription and DNA replication) contribute considerably to the DNA damage load in a cell. It is estimated that between one thousand and one million DNA lesion occur in every human cell per day (Lodish et al. 2004) (Fig. 2).
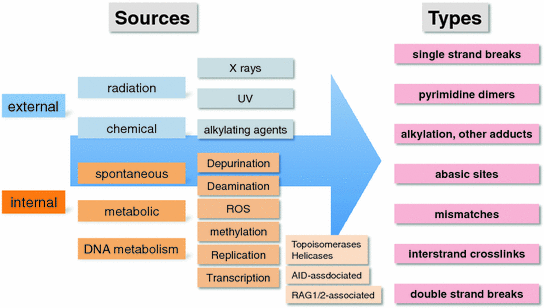
Fig. 2
Sources and types of DNA damage. Note that this is not an exhaustive list
It should be noted that a minimum of two DNA double-strand breaks (DSBs) is required for a chromosomal translocation to occur (Fig. 3). Certain cells, like developing B and T cells, will introduce directed DSBs into their genomes as part of the immunoglobulin or T cell receptor maturation process (Zhang et al. 2010). This process has been especially well studied both in terms of the mechanisms involved and in the context of its contribution to the formation of recurrent chromosomal translocations found in lymphoid malignancies.
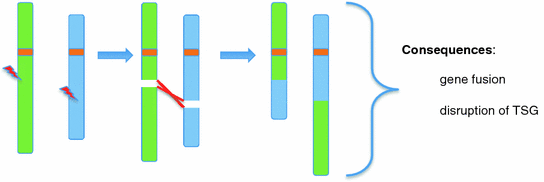
Fig. 3
Faulty repair of two DNA DSBs results in balanced chromosomal translation. The example shows two DSBs on two non-homologous chromosomes
DNA replication is a considerable source of DNA damage. DNA replication is an extremely complicated process which involves among many other steps the initiation of replication at specific origins of replication, the synthesis of two new DNA strands as a leading and a lagging strand and the ligation of the newly synthesized fragments (Okazaki fragments) of the lagging strands. At any of these steps problems can occur which might lead to abortive replication with replication fork stalling and possibly DNA strand breaks (Halazonetis et al. 2008). The fidelity with which the correct nucleotides are incorporated into the new DNA strands depends on the intracellular concentration of the individual deoxyribonucleotide triphosphates. Under certain conditions, like enhanced cellular proliferation, the local nucleotide pools can become depleted and nucleotides incorrectly incorporated at an increased rate (Bester et al. 2011). This in turn can lead to replication fork stalling, repair attempts, faulty repair and eventually also to DSBs, which might result in chromosomal rearrangements.
The most widely studied source of DSBs in the context of chromosomal translocations are the DNA breaks that are introduced by the RAG1/2 enzymes in the course of lymphoid cell differentiation (Zhang et al. 2010).
3.3 Types of DNA Damage and Their Source
As stated above, the most relevant type of DNA damage for the formation of a chromosomal translocation is a DSB. However, other types of DNA damage like abasic sites, base mismatches etc., can result in DSB if these lesions cannot be repaired properly or if the lesion (damaged nucleotide or strand cross-link) is encountered during DNA replication. We will briefly review these types of DNA lesions and then focus on DSBs (Fig. 2).
3.3.1 Uracil, Abasic Sites, 8-Oxoguanine, Single Strand Breaks, Pyrimidine Dimers, O-6-Methylguanine
Spontaneous deamination of a cytosine residue will lead to the generation of a uracil residue and the spontaneous depurination reaction will remove a guanine or adenine base from the DNA strand. Both, deamination and depurination will create an abasic site. It should be noted that these reactions occur spontaneously at an estimated rate of 5000 depurination and about 100 deamination reactions per human cell per day (De Bont and van Larebeke 2004).
Alkylating agents and oxygen radicals attack nucleotides at various places creating bulky DNA adducts or single stranded breaks, respectively. Oxygen radicals will also generate 8-oxoguanine. Alkylating agents are mostly of external origin while oxygen radicals can be derived both from external sources such as ionizing radiation and intracellular sources such as ROS generated by metabolic processes. Single strand DNA breaks are frequently generated due to radiation or ROS.
Photoproducts such as pyrimidine dimers are the result of direct UV radiation. They can lead to stalled replication forks and eventually to DSBs.
Guanine nucleotides are spontaneously methylated to O-6-methylguanine in a non-enzymatic reaction by the ubiquitous methyl group donor S-adenosyl-methionine (De Bont and van Larebeke 2004).
3.3.2 Replication Errors and Mismatches
DNA replication is a highly complex process and requires the interplay of many proteins that work together in the so-called replisome, which in eukaryotic cells contains at least three different DNA polymerases. While this process has a high fidelity, wrong nucleotides can be incorporated leading to mismatches. Replication through microsatellite repeats can easily lead to small insertions and deletions and the expansion or contraction of the repeat. Replication errors are more frequent when the intracellular nucleotide pool has become depleted as might be the case in a rapidly proliferating tumor (Bester et al. 2011).
The replisome also contains or attracts proteins that can detect replication errors and will then recruit repair proteins. Sometimes repair attempts are not successful and will result in DSBs (see below).
3.3.3 Double Strand Breaks (DSBs)
As mentioned above, DSBs, if not repaired faithfully, are the main source of chromosomal translocations. Several cell external and cell intrinsic causes for DSBs are well known. It is estimated that about 50 DSB occur every day in a normal human cell (Vilenchik and Knudson 2003) (Fig. 4).
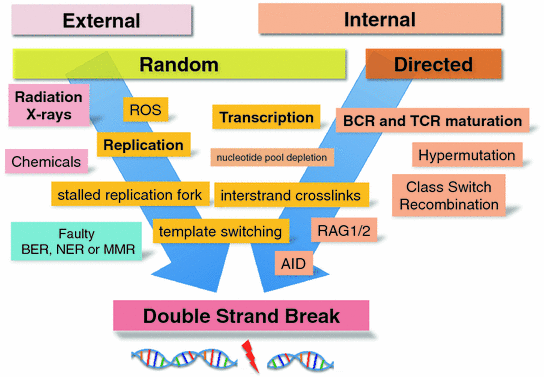
Fig. 4
Causes of DSBs. Abbreviations: ROS reactive oxygen species; BER base excision repair; NER nucleotide excision repair; MMR mismatch repair; AID activation-induced cytidine deaminase; RAG1/2 recombination associated genes 1 and 2; BCR B cell receptor; TCR T cell receptor
Random DSBs Due to External Influences
There are many external agents that can lead to DSBs in a cell. The most well known are high-energy radiation in the form of gamma rays, alpha particles or beta rays. All these high energy radiation sources will either directly shatter DNA or lead to the formation of an ionization trail with highly reactive molecules such as ROS, which will in turn attack and react with DNA causing DSBs. The most effective radiation in producing DSBs are alpha particles, which deposit a high amount of energy in the form of ionization events over a very short distance in the cell.
In addition to leading directly to a DSB as a consequence of the action of ROS or a direct shattering of the DNA strands, radiation will also generate interstrand crosslinks which will cause a problem during replication (see below).
Random DSBs Due to Internal Influences
ROS are normally produced in a cell as the result of metabolic processes (Sallmyr et al. 2008). If ROS are not scavenged efficiently or produced at a higher rate due to increased metabolic activity, the likelihood that DSB are generated increases.
DNA replication can be a major source of DSB breaks. The replication fork stalls if it encounters a mismatch or an intrastrand crosslink. Attempts to repair mismatches generated during replication can lead to abortive repair by the mismatch repair pathway which will lead to DSBs. As mentioned above, the frequency of mismatches is increased if the nucleotide pools are depleted due to rapid proliferation causing the DNA polymerases to incorrectly incorporate nucleotides at a higher rate (Bester et al. 2011).
Directed DSBs Due to Internal Processes (e.g., Immunoglobulin Rearrangements)
Interestingly, there are several physiological processes which introduce DSBs “on purpose”. These processes are required for the generation of genomic rearrangements at the immunoglobulin and the T cell receptor loci during the maturation of B and T cells, respectively (Dudley et al. 2005). The mechanisms that lead to these DSBs and their proper resolution in the form of productive rearrangements have been studied in great detail and their involvement in the generation of chromosomal translocations that drive lymphoid malignancies has been subject to intense scrutiny (Zhang et al. 2010).
RAG 1/2 Associated DSBs
The RAG1/2 recombination-associated endonucleases are responsible for introducing DSB to initiate the process of VDJ recombination at IGH, TRB, and TRD loci and the VJ recombination process at the IGK, IGL, TRA and TRG loci. The DSBs introduced by the RAGs are the first step in recombining one of many V (variable) segments with one of many J (joining) segments to create functional immunoglobulin or T cell receptor genes. In the case of the IGH, TRB and TRD loci a D (diversity) segment to J segment recombination precedes the V–J recombination for a full VDJ recombination event. The V(D)J recombination is completed by repairing or joining the DSBs with the classical nonhomologous end-joining pathway (C-NHEJ) (Zhang et al. 2010) (see below).
DSB are very dangerous for a cell because they compromise the integrity of its DNA. Since the purpose of these DSBs is to produce a functional gene, the RAG endonucleases do not cut randomly in the genome but are guided by specific recombination signal sequences (RSS), which consist of heptamer/nonamers that are separated either by a 12 bp or a 23 bp spacer (Tonegawa 1983). The RAGs only introduce DSB in two heptamer/nonamer sequences if one contains a 12 bp and the other a 23 bp spacer. Even though there is sequence specificity and several constraints in where DSBs can be introduced by the RAGs, some variations in the heptamer/nonamer sequences are tolerated. This means that DSBs can be introduced at other genomic loci that happen to have sequences which resemble the heptamer/nonamer sequences of the immunoglobulin and TCR loci. It is quite obvious that DSBs outside the immunoglobulin or TCR loci can lead to unintended and potentially dangerous chromosomal rearrangements if joined via the C-NEHJ pathway (Robbiani et al. 2009).
AID Associated DSBs
In addition to the diversity in the immunoglobulin and TCR genes generated by the V(D)J recombination process, more diversity is achieved by the process of somatic hypermutation (SHM). In SHM, mutations are introduced into the variable region exons of the immunoglobulin or TCR genes. The key enzyme responsible for SHM is the activation-induced cytidine deaminase (AID). AID deaminates cytidines in single-stranded DNA regions. The AID enzyme does not only introduce mutations into the variable region exons (Li et al. 2004) but is also responsible for the DSB required for class switch recombination (CSR) (Muramatsu et al. 2000). Since AID needs single-stranded DNA, its activity is coupled to active transcription which requires the unwinding and melting of the DNA double strand. Thus, AID is targeted by active transcription to the switch region of the constant region exons (for CSR) or to the variable region exons (for SHM). The cytidine deamination events introduced by AID into the switch regions of the constant region exons are processed to DSB through the activity of proteins involved in DNA repair (Chaudhuri and Alt 2004; Di Noia and Neuberger 2007). What causes AID induced deamination events to lead to single nucleotide mutations and small indels at the variable region exons on the one hand and to DSB in the switch regions of the constant region exons on the other hand is currently unknown (Muramatsu et al. 2007). High throughput genome-wide translocation sequencing showed that AID-associated DSBs that occur outside the normal loci for CSR are more frequent in actively transcribed regions (Chiarle et al. 2011).
DSB Due to Normal Chromatin Movement and Processes (Topoisomerase and Helicase Action)
The nucleus is a very crowded place occupied by 46 linear DNA molecules with a total length of about 2 m crammed into a small space of just a few cubic micrometers (approximately 100–200 µm3). Many processes in the nucleus like transcription, DNA replication and chromosome condensation require the unwinding of supercoiled DNA to relieve torsional stress and to allow replication and transcription factors to gain access to specific sites. Due to topological constraints it frequently becomes necessary to break one DNA double strand temporarily to pass another DNA strand through this gap. Introducing a DSB into a DNA molecule to pass another DNA strand through the gap and then repairing the gap is a very risky process and can easily lead to a faulty repair if another DSB is in the vicinity. Topoisomerases and helicases and a number of DNA repair proteins are involved in this process (Kaneko et al. 2004; Plank and Hsieh 2009; Vos et al. 2011).
To relieve torsional stress of a DNA molecule, e.g., during transcription, DNA helicases will introduce a nick in one strand of a dsDNA molecule, release the stress and then re-ligate the single-stranded nick. This process is also risky since it can result in a DSB (Carrasco et al. 2014).
3.4 Detecting DNA Damage
Any DNA damage in a cell has to be detected rapidly so that it can be repaired before the lesion is passed on to the daughter cells. One of the worst consequences of improperly DNA damage is cancer.
While many of the DNA lesions described above are detected by specific proteins and mechanisms, we will focus on the pathways in place to detect DSB, which are the predominant substrate for the generation of chromosomal translocations.
3.4.1 Detecting DSB and Signaling for Repair
It is absolutely crucial for cell survival to quickly detect any DSBs and repair them efficiently. A DSB generates two ends of a double stranded DNA molecule, which immediately attract the attention of several proteins. These proteins will then orchestrate the recruitment of additional DNA repair proteins. This is an extremely complicated process involving a great number of proteins, which is still under intense investigation. Several pathways and proteins that are working in this DSB surveillance network have been identified (Fig. 5).
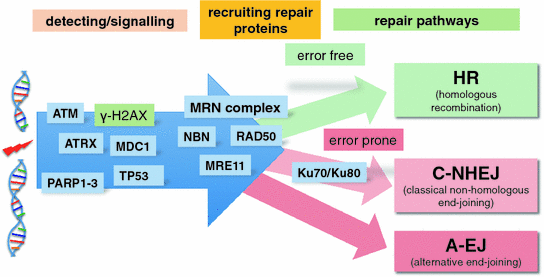
Fig. 5
Double strand breaks (DSBs): Detection, signalling, recruitment of repair proteins and repair pathways. The MRN complex consists of RAD50, NBN and MRE11. The repair pathways are divided into error free (green) and error prone (red)
Even though telomeres technically constitute ends of linear dsDNA molecules they are protected or hidden from the detection of the break surveillance proteins by their special structure and a large number of telomere-associated proteins (Slijepcevic and Al-Wahiby 2005).
Immediately after a DSB occurs, the surrounding chromatin undergoes extensive modifications which are accompanied by various posttranslational changes of histones and other proteins (poly(ADP-ribosyl)ation, ubiquitylation, sumoylation, acetylation, phosphorylation). These modifications also extend from the site of the DSB and lead to the recruitment of DNA repair proteins (Lukas et al. 2011).
Poly(ADP-ribosyl)ation
The first detectable event after a DSB is poly(ADP-ribosyl)ation (PAR) mediated by the PARP1-3 enzymes. PAR modifies lysine residues of the core histones tails. PAR of the histone tails leads to the recruitment of the chromatin remodelling complex NuRD/CHD4 and polycomb complexes (Chou et al. 2010).
Gamma H2AX (Phosphorylated Form of H2AX)
Another early event at a DSB is the phosphorylation of histone H2AX by ATM to form gamma-H2AX. Gamma-H2AX is recognized by its sensor MDC1 (mediator of DNA damage checkpoint protein1), which interacts among others, with the Nijmegen breakage syndrome protein, NBS1 (Goldberg et al. 2003). NBS1 in turn interacts with ATM and tethers it to the DSB to increase local gamma-H2AX concentration. MDC1 recruits a number of other proteins like RNF8 which subsequently leads to the recruitment of BCRA1, RAD18, PTIP (Pax transactivation domain interacting protein) and P53BP1 to the sites of DSBs (Lukas et al. 2011).
It should be noted that replication stress in the form of stalled replication forks also elicit gamma-H2AX formation via ATR (ATM and Rad 3-related) kinase activity (Wang et al. 2011).
TP53
The TP53 tumor suppressor gene plays an important role in the detection and signaling of DNA damage in a cell and orchestrating cellular responses to DNA damage like cell cycle control and apoptosis TP53 (Meek 2009).
53BP1
P53BP1 (P53 binding protein 1) can shield under-replicated DNA regions after the cell has gone through mitosis from the activity of DNA nucleases until the under-replicated DNA regions have been repaired. These under-replicated DNA regions, which are often found at common fragile sites, can become visible in mitosis as so-called ultrafine DNA bridges. 53BP1 nuclear bodies are found in the nuclei of cells that have just passed through mitosis (Lukas et al. 2011).
It should be noted that the exact molecular mechanisms that lead to DSB recognition are far from being completely understood and the description above offers only a glimpse of the complexity that is already known. These mechanisms also vary from cell type to cell type and also depend on the origin of the DSBs.
3.4.2 Signaling During Repair
Once the cell has detected one or more DSBs or other DNA lesions, it needs to repair the damage or make the decision that the damage is too extensive to be repaired, halt cell division or undergo programmed cell death. How this decision is made is not entirely clear. Rather than assessing the damage and making a ‘conscious’ decision that the damage is too extensive, the cell will attempt to repair the damage and while the repair process is in progress, the progression through the cell cycle will be blocked. Entering S phase, or even mitosis, with unrepaired DNA lesions can have catastrophic consequences.
Several well-known tumor suppressor genes function in pathways that prevent a cell from progressing through the cell cycle as long as there are unrepaired DNA lesions. These include the retinoblastoma gene (RB), the TP53 gene and the INK4A/B genes as well as proteins that are more directly involved in DNA repair (Huen and Chen 2010; Sperka et al. 2012).
3.5 Repairing DNA Damage
Just as there are many different types of DNA lesions, there are many damage repair pathways that are very specific for certain lesions. We will only briefly mention these specialized repair pathways and then focus on the pathways involved in the repair of DSBs. However, the failure to properly repair other types of DNA damage such as mismatches, uracil bases, abasic sites, pyrimidine dimers or chemically modified bases (alkyl groups, O-6-methylguanine) can also result in DSBs (Fig. 6).
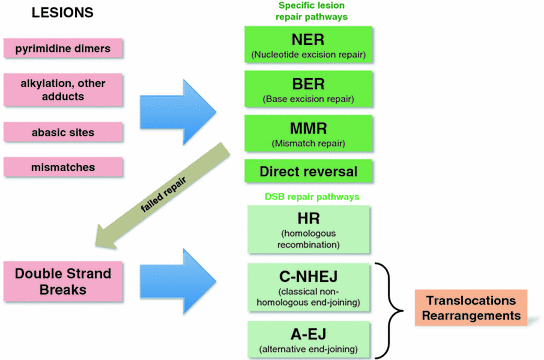
Fig. 6
Different types of DNA lesions and their repair pathways. Note that failed repair of a specific lesions might lead to a DSB which will then have to be repaired. Translocations and other rearrangements are mostly the result of C-NHEJ and A-EJ joining the incorrect DNA ends
3.5.1 Base Excision Repair (BER)
Abasic sites, uracil bases, 8-oxoguanine and single strand breaks (check ssBreak repair) are efficiently repaired by the base excision repair (BER) process (Krokan and Bjørås 2013). This process involves the removal of the uracil by DNA glycosylases and then the removal of the sugar phosphate by the action of the AP endonuclease (AP for apurinic and apyrimidinic site) and a phosphodiesterase. This creates a one nucleotide gap in the DNA strand with the lesion, which is then filled by the action of DNA polymerase(s) and sealed by a DNA ligase (Krokan and Bjørås 2013).
3.5.2 Nucleotide Excision Repair (NER)
Bulky DNA adducts and pyrimidine dimers and many other types of lesions are repaired by the nucleotide excision repair (NER) process. The altered DNA strand is recognized by a multi-protein complex that introduces single strand nicks on either side of the lesion. The nicks are spaced 12 nucleotides apart in bacteria but more than 24 nucleotides apart in eukaryotes. The intervening fragment is removed by the action of a helicase and the resulting single strand gap is repaired by the action of polymerases and finally sealed by ligases. Mutations in NER pathway proteins, especially those that are involved in the initial recognition of the damaged DNA, are found in Xeroderma pigmentosa patients (Shuck et al. 2008).
3.5.3 Direct Reversal
Certain DNA lesion like O-6-methylguanine and thymidine dimers can also be repaired by direct reversal mechanisms. The methyl group of O-6-methylguanine is removed by the action of MGMT (Tano et al. 1990), and UV-induced pyrimidine dimers can be reversed by the action of the photolyases (Kneuttinger et al. 2014). Photolyases, which catalyze the direct reversion of thymidine dimers, are not functional in placental mammals (Lucas-Lledó and Lynch 2009).
3.5.4 Mismatch Repair
Mismatches and smaller insertions and deletions that arise frequently during DNA replication are efficiently repaired by the mismatch repair process (MMR). A multiprotein complex, containing the components of the MMR machinery and additional proteins, is loaded onto the freshly replicated DNA double strands, scans the DNA and recognizes distortions of the helical structure caused by base mismatches between the two strands. Once a distortion is detected the newly synthesized DNA strand is nicked by the endonuclease in the complex and the endonuclease exo1 removes several nucleotides including the mismatches in this strand. Then the gap in the strand is filled by the action of DNA polymerase delta and the repair is completed by the action of a DNA ligase (Li 2008). There are about 10 proteins involved in MMR and mutations in several of the genes coding for these proteins are found in hereditary cancer syndromes like HNPCC (Müller and Fishel 2002) (Fig. 6).
3.5.5 Double Strand Break Repair
Faulty repair in any of the repair pathways described above can result in DSBs, which can lead to chromosomal translocations. The cell will attempt to repair any DSB using one of three or four DSB repair pathways. DSB repair pathways can be subdivided into error-prone and error-free pathways. In this context, error-free has to be understood as theoretically error free or usually error free. After an error free repair, the nucleotide sequence of the repaired region will be identical to the nucleotide sequence before the DSB.
Homologous Recombination (HR)
Homologous recombination is a usually error free DSB repair pathway. This process operates in the S and G2 phase of the cell cycle because it requires an intact sister chromatid, which serves as the template for the DNA repair. The MRE11/RAD50/NBS protein complex is loaded onto the end of the DSBs where the 5′-3′ exonuclease activity of MRE11 removes nucleotides from one of the strands of the free dsDNA end (Daley et al. 2013; Lammens et al. 2011; Williams et al. 2010). This leads to an exposed single-stranded 3′ DNA overhang, which is processed with the help of replication proteins A (RPA), RAD51, BRCA2 and several other proteins into the RAD51-ssDNA-nucleoprotein filament. The RAD51-ssDNA-nucleoprotein filament will then invade the dsDNA of the homologous site on the sister chromatid, which serves as a template for the error-free synthesis of the DNA across the DSB region (Pellegrini et al. 2002; Popp and Bohlander 2010; Yang et al. 2002). After the Holliday junctions are resolved, the error-free repair is completed. This repair pathway can result in sister chromatid exchange. The DSB repair by HR is a sterically very complicated process and involves, in addition to the proteins mentioned above, several other important proteins like RECQL2 (Werner Syndrome), BLM (RECQL3) (Bloom Syndrome), BRCA2 (familial breast cancer), RAD54, PALB2 (familial pancreatic cancer), FANCM, FANCC (Fanconi anemia) and others (Daley et al. 2013; Popp and Bohlander 2010). A number of these proteins are mutated in hereditary tumor or genome instability syndromes and are also somatically mutated in these tumors (Bunting and Nussenzweig 2013; Ellis et al. 1995; Jones et al. 2009; Meetei et al. 2003; Strathdee et al. 1992; Wooster et al. 1995; Yu et al. 1996).
Single Strand Annealing (SSA)
BIR (Breakage Induced Replication Template Switching)
Another, mechanism to generate a translocation would be for the DNA replication machinery to switch to another chromosome as a template. This so-called breakage-induced replication (BIR) template switching can be induced by a break during DNA replication (Bunting and Nussenzweig 2013).
Classical Non-Homologous End Joining (C-NHEJ)
The C-NHEJ repair pathway for DSB is error-prone, which means that after the joining of the broken ends there will be, mostly smaller, deletions or insertions at the location of the former DSB (Rassool 2003; Roth and Wilson 1986). This process does not need an intact template strand from the sister chromatid and is therefore the major repair pathway for DSBs during the G1 and S phases of the cell cycle. The C-NHEJ pathway is also far more likely to join DSBs that do not belong together since it does not require homologous sequences to guide the repair process. It is, together with alternative end joining (A-EJ), the major repair pathway for removing DSB in the cells. C-NHEJ is used in V(D)J recombination at the immunoglobulin and T cell receptor loci for repairing the breaks that are generated in the process of somatic recombination and CSR.
To initiate the C-NHEJ process, the DSB DNA ends are bound by the KU70/80 proteins to prevent the drifting apart of the DSB ends (Soutoglou et al. 2007). Then DNA-PKCs and the MRN complex (MRE11-RAD50-NBS) are recruited to the breaks. The MRN complex is crucial in all three DSB repair processes (HR, C-NHEJ and A-EJ). It executes and coordinates many of the activities that are required for DSB repair. For example, the RAD50 coiled-coil domains extend from the core MRN complex which occupies the DSB end to connect the DSB to the other break or the sister chromatid (Lammens et al. 2011).
The MRN complex is responsible for some resection at the DSB and then the broken DNA ends are joined together by the activity of the DNA ligase IV-XRCC4 complex (Roth and Wilson 1986).
Other important proteins involved in C-NHEJ are Artemis (Moshous et al. 2001) and Cerunnos (XLF) (Ahnesorg et al. 2006; Buck et al. 2006). Both proteins were identified because mutations in these factors lead to certain severe combined immunodeficiency syndromes, in which immunoglobulin recombination and TCR recombination are severely compromised.
Alternative End Joining Pathway (A-EJ) or Microhomology-Mediated End-Joining (MMEJ)
There are probably several A-EJ pathways. A-EJ is the repair of DSB in the absence of KU70/80, XRCC4 or Ligase 4 (Daley and Wilson 2005; Wang et al. 2003). The A-EJ pathway also relies on the MRN complex as the crucial component of the repair process (Popp and Bohlander 2010).
A-EJ repaired DSBs are characterized by the presence of stretches of microhomologies of six to eight base pairs. In some models, A-EJ pathways are responsible for the majority of DSB repairs that resulted in chromosomal translocations (Boboila et al. 2010; Simsek and Jasin 2010).
In systems with inducible chromosomal translocations, the analysis of the breakpoints showed that microhomology-based mechanisms were responsible for only a minority of de novo translocations (Daley and Wilson 2005; Wang et al. 2003). Next generation sequencing studies also showed that less than a third of human germ line chromosomal translocations showed microhomologies at the breakpoints. The breakpoints showed more complicated structures with fragmentation of local DNA sequences, small inversions and deletions. This suggests that C-NHEJ rather than A-EJ contributes to a substantial fraction of human germline chromosomal translocations (Chiang et al. 2012).
4 Translocations
4.1 Translocations—the Tip of the Iceberg
It should be noted that there is now solid evidence that translocations between non-homologous chromosomes are rare compared to intrachromosomal rearrangements (Mahowald et al. 2009; Zhang et al. 2012) between loci that are closer to each other in the interphase nucleus than loci on different chromosomes (Campbell et al. 2008; Pleasance et al. 2010a, b; Stephens et al. 2009).
In addition to the classical, balanced translocation (like the t(9;22)(q34;q11) or the t(8;14)(q24;q32) (Dalla-Favera et al. 1982; Rowley 1973; Taub et al. 1982)), two DSBs can lead to a great variety of other chromosomal rearrangements (Fig. 7).
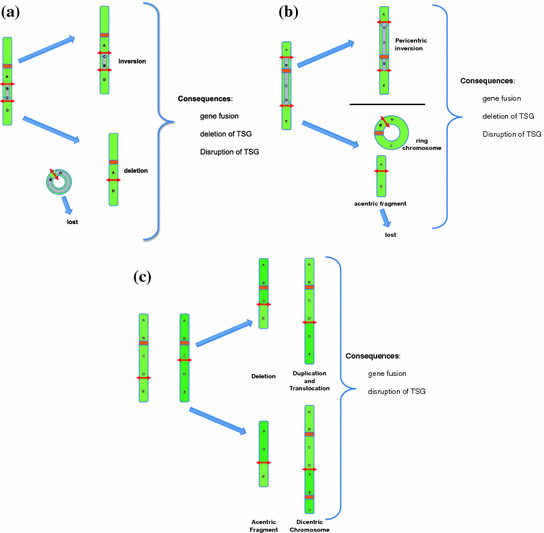
Fig. 7
Possible types of rearrangements after two DSBs depending on the location of the DSB. Note that not all possible constellations of two DSBs are shown. The case of two DSBs on two different chromosomes is shown in Fig. 3. Ring chromosomes and dicentric chromosomes, as well as acentric fragments are frequently mitotically unstable and lead to aneuploidies. a Two DSBs located on the same arm of the same chromosome. This can result in a paracentric inversion or a deletion. The acentric circular fragment will be lost during mitosis. The consequences of such rearrangements can be a gene fusion event (resulting in a fusion gene or deregulated gene expression) and/or the disruption or deletion of a gene (tumor suppressor gene). b Two DSBs located on different arms of the same chromosome. This will result in a pericentric inversion or in a ring chromosome and an acentric fragment which will be lost during mitosis. c Two DSBs located on the same arms of two homologous chromosomes. The result is either a deletion accompanied by a duplication or a dicentric chromosome and an acentric fragment
4.2 DSBs and the Resulting Chromosomal Rearrangements
Depending on the number of co-occurring DSBs and whether the DSBs occur on the same chromosome arm, on different chromosome arms of the same chromosome or on different chromosomes, different chromosomal rearrangements will result. It is quite obvious that the complexity and types of rearrangements increases rapidly with increasing number of co-occurring DSBs. We will only discuss a few scenarios to illustrate the increasing complexity of possible rearrangements as the number of DSB increases and that the most common rearrangements following DSBs have only become “visible” through new technical developments in sequencing, which allowed the analysis of whole tumor genomes at unprecedented resolution.
4.2.1 Consequences of Two DSBs
If two DSBs occur on the same arm of a chromosome there are four free dsDNA ends that can either be repaired in their original order or be joined in a different order, which will result in genomic rearrangements. There are only two possible outcomes of this non-correct joining: (1) Deletion: If the ends of the chromosome fragment between the two breaks are joined to form a circular DNA molecule and the two ends of the flanking fragments are joined, an interstitial deletion will be the outcome. Since the circle does not have a centromere, it will be lost in subsequent cell divisions. (2) Inversion: The fragment between the two breakpoints can be inverted resulting in a paracentric inversion (Fig. 7a).
If the two DSBs occur in different arms of the same chromosome and are joined in the incorrect order there are also only two outcomes but the consequences are more complicated. (1) If the ends of the fragment containing the centromere are rejoined, a ring chromosome will be produced. The fragments containing the telomeres might then join to form an acentric chromosome, which will be lost in subsequent cell divisions. The net result will be the deletion of the DNA sequences distal of the two breakpoints (i.e., the sequences towards the telomeres). (2) The alternative, incorrect order of joining the four DSB break ends will result in a pericentric inversion of the chromosome (Fig. 7b).
If two DSBs occur on the same arm of two homologous chromosomes and are joined incorrectly, translocations, interstitial duplications and deletions will result. Alternatively, an acentric fragment and a dicentric chromosome can form. Both are mitotically very unstable (Fig. 7c).
Finally, if two DSBs occur on different, non-homologous chromosomes and are joined in the incorrect order, two possible outcomes are observed:
1.
A balanced chromosomal translocation occurs if the DSB ends are joined in the correct orientation with respect to their centromere to telomere orientation (Fig. 3). The ends on the fragments with the centromeres need to be joined to the ends on the fragments with the telomeres.
2.
A dicentric chromosome and an acentric fragment will be generated if the two ends connected to a centromere and the two ends connected to the telomeres are joined, respectively. Dicentric chromosomes are extremely unstable during cell division and are subject to additional breakage events while the acentric fragments will be lost during mitosis (see Fig. 7c, lower right half).
4.2.2 Consequences of Three and More DSBs
The number and types of possible rearrangements that can result if the ends from three DSB breaks are joined in an incorrect order is quite large. Three DSBs will generate six DNA ends. These six DNA ends can theoretically only be joined in three non-correct ways. However, the type of rearrangements that can result from three DSBs also depends on where these DSBs are located with respect to each other. For example, if the breaks are all on the same arm of the same chromosome the possible rearrangements are different from the situation in which all three DSBs are on different, non-homologous chromosomes. For example, if all three DSBs are on the same chromosomal arm, there can either be a deletion or an inversion rearrangement. If the three breaks occur all on different chromosomes, a three way balanced translocation can be one of the results. If two breaks occur on the same chromosome arm and the third break on a different chromosome, interchromosomal insertions will be observed.
If the number of DSB breaks increases, the number of possible rearrangements will increase in a non-linear fashion. The most extreme example of rearrangements resulting from multiple DSBs is a so-called ‘chromothripsis’ event. The complexity of chromothripsis events has only recently been discovered and required next generation sequencing approaches to decipher (Molenaar et al. 2012; Stephens et al. 2011). It should be noted that the exact causes of chromothripsis events are not entirely understood at the moment. One of the explanations would be a catastrophic event that produces many, local DSBs which are then repaired in the wrong order. There is also the possibility that locally disrupted DNA replication leads to a chain of microhomology-mediated template switching events (see below) (Crasta et al. 2012; Holland and Cleveland 2012) .
4.3 Steric Constraints on the Formation of Chromosomal Rearrangements
4.3.1 Arrangement and Mobility of Chromosomes in the Interphase Nucleus (Chromosome Territories)
In the interphase nucleus, chromosomes are not arranged randomly like spaghetti on a plate. Each chromosome occupies a so-called chromosome territory (CT) (Cremer and Cremer 2001) (Fig. 8). Chromosome territories are of complex shapes. Adjacent chromosome territories interdigitate with each other (Cremer and Cremer 2001). There is no fixed order of how CTs are arranged with respect to each other in the interphase nuclei of a given cell type (i.e., there is no rule that the CT of chromosome 1 should always make contact with the CT of chromosome 10, for example). The arrangement of CTs with respect to each other seems to be dynamic and also changes during cellular differentiation. There is a tendency of CTs of chromosomes with a high gene density to be located more centrally in the nucleus and the CTs of gene poor chromosomes to be found more frequently in the nuclear periphery (Cremer et al. 2001).
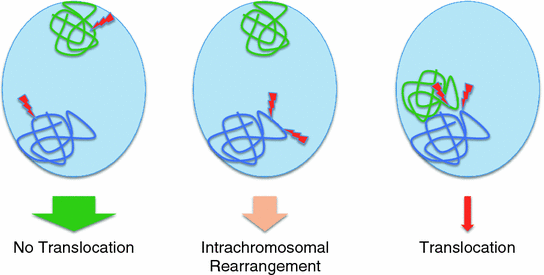
Fig. 8
Possible scenarios for the location of two simultaneously occurring DSBs in the interphase nucleus and the likely outcome. The width of the arrows correlates with the likelihood of the different outcomes. Only two chromosome territories (CTs) are shown as example. Left the two DSBs occur in different CTs which are far apart in the interphase nucleus. Middle two DSBs occur in the same chromatin domain on the same CT. Right two DSBs occur in different, adjacent CTs but closely spaced in intermingling chromosome domains
Interphase DNA is wrapped around an octamer of core histones which form the nucleosomes. The DNA wrapped around individual nucleosomes has the appearance of bead on a string and is the basic 10 nm chromatin fiber (Belmont 2006; Misteli 2010). In the interphase nucleus, the chromatin fiber is further compacted with linker histones and non-histone proteins and organized into so-called chromatin domains (CDs), containing on the order of 1 Mbp of DNA. CDs form the basic units of higher-order chromatin organization (de Graaf and van Steensel 2013). While the interphase chromatin is not held in place by a rigid matrix, there is evidence that the movement of the chromatin, and hence the DNA, is spatially confined. Thus, the two ends of a DSB are not free to move to any place within the nucleus but have been shown to move within a radius of approximately 1 µm (Soutoglou et al. 2007).
These steric constraints, namely the packaging of chromosomes in CT, the organization of the DNA into CD, and the resulting limited mobility of DSB ends have very important implications for the frequency and types of chromosomal rearrangements that can occur as a consequence of DSBs.
As stated above, a minimum of two DSBs is required for a chromosomal rearrangement to occur. In addition, these two DSBs have to occur at the same time. There are an estimated 50 DSBs per nucleus per day (Vilenchik and Knudson 2003). However, DSB repair is usually initiated within a few minutes of the breakage event. At that time, the broken ends are still very close together and have not moved apart very far because of the limited time available and the constraints imposed on the DNA by the higher order chromatin organization (Soutoglou et al. 2007). In order to rejoin the ends of one DSB (DSB-A) with the ends of another DSB (DSB-B), DSB-A and DSB-B have to occur close enough to each other in the interphase nucleus and at almost the same time. Even if two DSBs occur at the same time in the same nucleus but are separated by more than 1 µm, it is unlikely that the DNA ends can move far enough so that a rearrangement can occur (Fig. 8).
Considering these limitations, it is quite safe to assume that chromosomal rearrangements have a much higher chance to occur if the two originating DSBs occur in close spatial and temporal proximity. Therefore, it is quite obvious, considering the arrangement of DNA in the interphase nucleus, that two DSBs that are close enough to each other to lead to a rearrangement are much more likely to be located on the same chromosome and also within the same or neighboring CD. These considerations imply that intrachromosomal rearrangements that involve breaks that are less than 1 Mbp (DSBs in the same CD) or only a few Mbp apart (DSBs in neighboring CDs) are much more frequent than interchromosomal rearrangements (i.e., translocations). Data from recent high throughput sequencing experiments of tumor genomes are supporting these assumptions. About 80 % of all the rearrangements found in these studies were indeed intrachromosomal, with the majority involving breakpoints that were just a few Mbp apart (Wijchers and de Laat 2011) (Fig. 8).
4.4 Functional Consequences of Translocations
Before we ask the question of why specific translocations recur, we will briefly consider the functional consequences of genomic rearrangements that result from DSBs. For the sake of simplicity, we will focus on the functional consequences of translocations, but the same principles do apply to deletions, inversions and more complicated rearrangements.
Related posts:
 Modelling: What Have Mouse Models for Chromosome Instability Taught Us?
Modelling: What Have Mouse Models for Chromosome Instability Taught Us?
 of Chromosomal Instability
of Chromosomal Instability
 of Aneuploidy in Cancer: Transcriptome and Beyond
of Aneuploidy in Cancer: Transcriptome and Beyond
 Diverse Effects of Complex Chromosome Rearrangements and Chromothripsis in Cancer Development
Diverse Effects of Complex Chromosome Rearrangements and Chromothripsis in Cancer Development
 of Chromosomal Aberrations in Solid Tumors
of Chromosomal Aberrations in Solid Tumors
 Dysfunction, Chromosomal Instability and Cancer
Dysfunction, Chromosomal Instability and Cancer
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


