Keywords
Reovirus, chemotherapeutics, radiotherapy, cancer, clinical trials
Introduction
The use of viruses in the treatment of cancer resulted from the observation that clinical remission was occasionally seen in patients who contracted an infectious disease. Early reports were almost exclusively related to hematological malignancies, which are associated with significant immune suppression. As early as 1904, there were reports of the beneficial effects seen in patients with leukemia who developed influenza . Although there were no reports of prolonged response (i.e., “cure”), even at this early stage there was anticipation that viruses may hold the key to a novel treatment long before the advent of conventional chemotherapy. More recent clinical reports have described the regression of leukemia, Hodgkin’s disease, and Burkitt’s lymphoma in association with measles infection . From these reports, investigators have deduced the following: Certain viruses can destroy cancer cells without causing undue harm to the patient, this is most often seen in patients with a suppressed immune system, and these virally induced remissions are short-lived and incomplete .
The concept of agents that could pass through filters (unlike bacteria) was first reported in 1898 by Martinus Beijerinck regarding tobacco mosaic virus . He defined the virus as a contagium vivium fluidum , but it was not until 1917, when the plaque assay was first introduced, that the particulate nature of viruses was ascertained . Electron microscopic images of viruses were obtained in 1939, allowing their structure to be observed for the first time . Our understanding of viruses increased considerably with the ability to grow viruses in cell culture in the 1950s . Since then, viruses have been studied in enormous depth: Their genomes have been sequenced, their physical structures defined, and their methods of replication and pathogenetic strategies elucidated. More recently, methods have been developed to genetically modify viruses to improve their capability as anticancer agents.
Methods to limit the pathogenicity of oncolytic viruses were attempted as early as the 1950s. The concept of viral adaptation, whereby the oncolytic potential of a virus was enhanced by numerous passages in a given tumor, had been demonstrated by Moore et al . . They rightly assumed that viruses would develop mutations that would enhance replication in the cells in which they were propagated. However, it was not until the advent of recombinant DNA technology in the early 1990s that the prospect of altering the selectivity of viruses through genetic engineering became possible. The first study to use this approach was published in 1991 by Martuza et al ., who used a genetically modified herpes simplex virus (HSV) . HSV mutants that lack thymidine kinase are only able to replicate in dividing cells. When these modified viruses were used in a nude mouse glioma model, they caused tumor growth inhibition and improved survival.
Since then, a large number of human pathogens have been genetically modified to increase their specificity for cancer cells and enhance their oncolytic capability. One of the most extensively studied and clinically tested to date is oncolytic reovirus.
Reovirus
Introduction
Reovirus is a wild-type “prototype” oncolytic virus currently being investigated as a novel therapy for cancer. This review gives a broad outline of reovirus biology and preclinical data obtained to date, and it discusses the oncolytic capability and features of reovirus that make it particularly attractive as a therapeutic agent. The clinical evaluation of reovirus is being led by centers in the United Kingdom and the United States. Past and current studies are described in this chapter.
Reovirus is a member of the family Reoviridae, which includes six genera, three of which infect animals: rotavirus, orbivirus, and reovirus . The name is a derivation of r espiratory e nteric o rphan viruses, acknowledging that they can infect the respiratory and gastrointestinal system but are not associated with any known disease. Reovirus is generally regarded as benign but can infrequently be associated with upper respiratory symptoms resembling a flu-like illness or mild gastrointestinal symptoms such as diarrhea. Reovirus is ubiquitous and has been isolated from untreated sewage, stagnant water, and rivers worldwide . Exposure to reovirus is very common in the human population, and up to 100% of human adults are seropositive .
Reovirus Structure
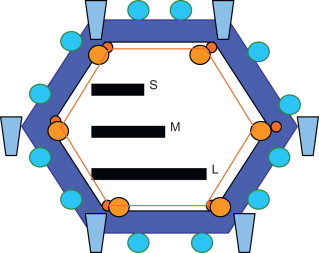
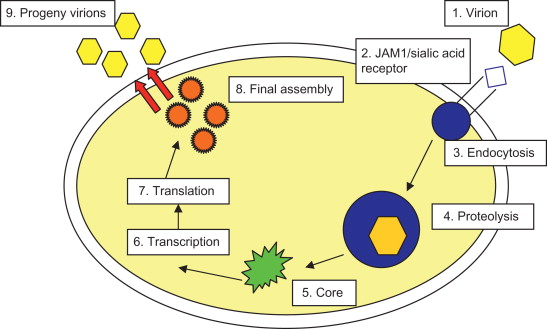
All members of the Reoviridae family have similar morphology. Reovirus virions consist of a nonenveloped, icosahedral capsid with a double shell of proteins. The genome is composed of double-stranded RNA (dsRNA), which is split into 10 segments of three sizes, designated L, M, and S. A simplified representation of the structure of reovirus is given in Figure 13.1 . A schematic representation of reoviral replication is shown in Figure 13.2 .
Reoviral Oncolysis
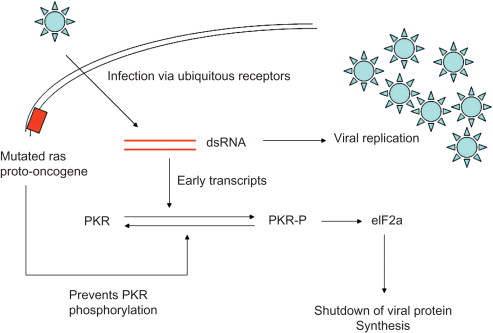
The oncolytic capability of reovirus was first recognized in the 1970s when wild-type reovirus was shown to replicate in transformed cells but not in normal cells. Cells expressing high levels of epidermal growth factor receptor (EGFR) and v-erbB (a truncated mutant of EGFR) were susceptible to reovirus infection , suggesting an intracellular factor associated with EGFR that was permitting reoviral replication . Further work identified the Ras pathway, a downstream signaling pathway from EGFR, as being crucial to sensitizing cells to reovirus . The Ras protein is a small G protein that is activated when guanosine triphosphate is phosphorylated, which in turn leads to the activation of downstream pathways that play an important role in cellular differentiation, proliferation, and motility and together can act synergistically to promote tumorigenesis. Ras can become activated either via mutation of the protein or via overexpression of the upstream EGFR or other receptor tyrosine kinases. Ras-activating mutations are present in 30–40% of all human tumors .
Reoviral infection of normal cells leads to the activation of the dsRNA-activated protein kinase (PKR). PKR is a serine/threonine protein kinase that requires dsRNA binding and phosphorylation to become activated . The main role of PKR is to defend cells against viral infection and to contribute to the antiproliferative response of interferon following viral infection. However, in cells harboring an activated Ras mutation, PKR is not phosphorylated and remains in its inactive state. Reoviral protein synthesis continues unchecked, the virus replicates within the cells, and cell lysis occurs . This mechanism is illustrated in Figure 13.3 .
Reoviral Therapy: Preclinical Data
More than 80% of cell lines originating from virtually all common tumor types have been found to be susceptible to reoviral oncolysis . Intratumoral injection of reovirus causes regression of syngeneic and xenograft tumors in immune-competent and -deficient animal models, respectively. Human glioma xenografts regressed by 80% after a single intratumoral injection of reovirus . Similar efficacy has been demonstrated in breast , lymphoma , and colon and ovarian cancer xenograft models.
Because the best understood mechanism of selectivity of reovirus replication for malignant cells was its relationship with Ras activation, early preclinical work focused on models involving tumors with activated Ras, including pancreatic (exocrine) adenocarcinoma , malignant glioma , medulloblastoma , and breast adenocarcinoma . However, subsequent studies have shown that activating Ras mutations are not a prerequisite for reoviral cytotoxicity because some cell lines with wild-type Ras are also susceptible . The mechanism of selectivity in these cells is not known. There is accumulating evidence that transcription regulation via PKR inactivation may not be the only mechanism for the preferential replication of reovirus in Ras-transformed cells. Two studies have shown that virus uncoating, assembly, and apoptosis-dependent release are enhanced in Ras-transformed cells . Whether any of these factors play a role in cells without an activated Ras pathway has not been determined.
Antiviral Response
One of the key factors limiting the potential of viral therapy for cancer is the induction of a rapid and potent antiviral humoral response. Attenuating this response has long been seen as a vital component in the development of this class of antitumor agent. Studies have shown early in vivo responses to systemic reovirus in both subcutaneous and metastatic models, only for the tumor to resume growth after several weeks despite continued therapy. This correlates with the development of a measurable antibody response mounted against reovirus . Combining intravenous reovirus treatment with immune suppression using cyclosporine A or anti-CD4/anti-CD8 antibodies resulted in further reduction in tumor size and a considerable prolongation in survival compared with viral therapy alone. Similarly, cyclophosphamide was also successful in modulating a neutralizing antibody response to reovirus when administered in a very specific (metronomic) dosing regimen . However, there are obvious risks with deliberate attenuation of the natural protective immune responses to reoviral infection with respect to noncancer tissue. Although coadministration of reovirus and cyclophosphamide was associated with improved efficacy, new toxicities were evident by the observation of increased viral replication in multiple organs and cardiomyopathy. By altering the schedule using metronomic dosing of cyclophosphamide (three injections separated by 6 days), significantly greater antitumor effects were demonstrated compared to the use of either agent alone, with minimal replication in essential organs. This highlights the “window of opportunity” with respect to combined immune modulation and reovirus treatments: a careful reduction rather than abrogation of humoral response. The current REO12 study is testing this hypothesis: Reovirus and increasing doses of cyclophosphamide are being administered, and the novel endpoint of the study is a demonstration of attenuation of the anti-reoviral antibody response.
In a study by Adair et al . , it was shown that despite the presence of neutralizing antibodies before viral infusion in all patients, replication-competent reovirus that retained cytotoxicity was recovered from blood cells but not plasma, suggesting that transport by cells could protect virus for potential delivery to tumor.
Given the potential risk of reovirus-induced toxicity in immunocompromised hosts, some authors have suggested an alternative strategy that would modify wild-type reovirus to improve selectivity. Due to its unique genetic composition, reovirus has not, until recently, been amenable to standard genetic modification. However, adaptation of reovirus in persistently infected cultured cells has been seen, and these attenuated viruses appear to be less pathogenic to nonmalignant cells while retaining their oncolytic potential . A novel reverse genetics system for dsRNA viruses has recently been described and may allow further research on reovirus attenuation .
Reovirus and Antitumor Immunity
In addition to the potential to inhibit the efficacy of oncolytic viruses through the production of antiviral antibodies, the immune system may augment therapy by bystander immune activation. Reovirus infection of tumor cells can enhance recognition by the immune system and help prime an antitumor response. The release of tumor-associated antigens in the context of reovirus-induced cell death can facilitate coincident innate immunity and subsequent specific priming against tumor-associated antigens.
Reovirus infection of melanoma cells has been shown to result in the release of proinflammatory cytokines, including interleukin (IL)-1, IL-6, IL-8, RANTES, eotaxin, MIP-1α, and MIP-1β . It also reduces the production of the immunosuppressive cytokine IL-10. This inflammatory response has been shown to cause bystander toxicity against reovirus-resistant tumor cells in vitro , thus providing another mechanism by which reovirus may exert its cytotoxic effects. The release of these inflammatory mediators acts to recruit other cells of the immune system to the tumor microenvironment. Reovirus directly activates dendritic cells, key regulators of the innate and adaptive immune response . These reovirus-activated dendritic cells then support innate killing by natural killer (NK) cells and also T cells. Supporting this concept is the finding that reovirus infection in both a murine melanoma model and human melanoma cells in vitro can generate adaptive antitumor immune responses .
In prostate cancer cells infected with reovirus, a pattern of increased proinflammatory cytokines and major histocompatibility complex class I molecule expression has been seen . Gujar et al . used an immunocompetent transgenic adenocarcinoma of mouse prostate (TRAMP) model to show that reovirus treatment induces the homing of CD8 + T and NK cells to a tumor and the display of tumor-associated antigens on antigen-presenting cells . This immunological activity led to the development of a strong anti-prostate cancer T cell response, which restricted the growth of subsequently implanted syngeneic tumor in an antigen-specific but reovirus-independent manner.
In a murine melanoma model using the reovirus-resistant B16ova cell line that expresses low levels of JAM-1, a single dose of reovirus-loaded T cells was able to purge lymph node and splenic metastases . In mice with severe combined immunodeficiency, reovirus failed to reduce tumor size in B16ova or reovirus-sensitive metastases, showing that an intact immune system is required for in vivo efficacy in this model. The same authors also showed that neither direct reovirus-induced oncolysis nor reovirus replication was required for the generation of antitumor immunity in an in vitro human system. Furthermore, the cytokine and chemokine response to reovirus infection appears to be PKR/NF-κB mediated and is able to prime the innate and adaptive antitumor activity even after live virus has been removed .
Taken together, these data imply a critical role for the immune system in mediating the efficacy of reovirus therapy. As previously stated, this may seem at odds with the finding that tumor response can be enhanced by combining reovirus with immunosuppressive agents such as cyclophosphamide. Although the improvement in viral delivery achieved by abrogating the viral antibody response is likely to be important for intravenous delivery of reovirus and the potential for increased persistence at the tumor site, this does not preclude the importance of the immune-mediated antitumor responses. It is likely that there is a fine balance between these factors. A strategy that could enhance antitumor immunity while improving delivery of the virus has yet to be determined.
Combination Therapy
Although reovirus alone has shown impressive antitumor effects in tumor models, its future potential as a mainstream cancer therapeutic is likely to be in combination with other treatment modalities.
Combination with Radiotherapy
Ionizing radiation is an important treatment for a range of cancers. However, it is clear that the Ras pathway play’s an important role in determining the sensitivity of tumors to radiotherapy . Overexpression of activating mutations of Ras has been associated with radiation resistance in vitro . Therefore, combining radiation with reoviral therapy that targets Ras-activated cells seems to be a logical route to take. Twigger et al . were the first to test combination treatments of radiation and reovirus both in vitro and in vivo . This group found that on a range of cell lines including head and neck, melanoma, breast, sarcoma, and adenocarcinoma, the effect was synergistic, and this was reflected in in vivo studies . The group hypothesized a number of mechanisms for the synergy. First, cells with mutated Ras that are radioresistant will be sensitive to reovirus. Thus, the two different treatments can target these two cell populations that may exist within the same tumor. Second, the stress response in irradiated cells may enhance reoviral replication. Third, radiation may increase the ability of reovirus to infect cells by interfering with cell surface receptors. Fourth, radiation changes in cell cycle may make tumor cells more permissive to reovirus infection. Finally, in vivo radiation may reduce the high interstitial pressure that may be a barrier to effective tumor spread. Despite these theories, no increase in viral output was evident when the tumor cells were irradiated before or after infection. Instead, the increased cytotoxicity seemed to be due to a marked increase in apoptosis with the combination treatment. They also found that radiation redistributes the cells toward G 2 /M and reovirus toward G 2 /M and S. However, with the two agents combined, this profile was overshadowed by the presence of a significant sub-G 1 population. This suggests that the combined treatment caused increased levels of apoptosis and that there seems to be no evidence that radiation actually makes the cell more permissive to reovirus.
Combination with Chemotherapy
A combination of chemotherapy and reovirus has resulted in enhanced cytotoxicity in a wide range of cancer cell lines, as shown by isobolographic analysis and tumor growth delay in in vivo models . Although the mechanisms of cell death when chemotherapy and reovirus are coadministered are likely to be different depending on the cell line and chemotherapeutic agent used, an increase in apoptotic cells was seen with combination compared to reovirus alone. With the combination of cisplatin and reovirus, there was evidence of enhanced viral replication compared to reovirus alone, but only within the tumor and not in distant organs . The addition of cisplatin had no effect on the humoral response to reovirus in a murine in vivo model, but it did abrogate the inflammatory cytokine response .
It has also been observed that there is increased sensitivity of tumor cells to reovirus during S and G 2 /M phases of the cell . Cells pretreated with hydroxyurea, or thymidine, were more sensitive to reovirus, particularly in S and G 2 /M phases, and there was evidence of increased viral replication. This finding was confirmed in vivo when hydroxyurea was given prior to intratumoral reovirus .
The combination of reovirus with multiple chemotherapy agents in human non-small-cell lung cancer (NSCLC) cell lines was reported by Sei et al . . The combined effects of reovirus with cisplatin, gemcitabine, or vinblastine demonstrated strong synergistic effects on cell killing but only in cell lines that were sensitive to these compounds. However, the combination of reovirus and paclitaxel was synergistic regardless of the cells’ sensitivity to either agent. The microtubule-stabilizing activity of paclitaxel was initially thought to have enhanced reoviral replication and thus may have been responsible for a more efficient and synergistic oncolytic effect. However, increased progeny outcome was also noted in the other drug combinations, which were not synergistic but, rather, in some cases antagonistic. Treatment of NSCLC cell lines with paclitaxel and virus resulted in increased poly(ADP-ribose) polymerase cleavage and caspase activity compared to single therapy, indicating that enhanced apoptosis, thought to be triggered by prolonged mitotic arrest, was most likely the reason for the synergistic effect. A study carried out by Heinemann et al . on the prostate cancer cell line PC3 also identified a synergistic effect with the taxane docetaxel and reovirus both in vitro and in in vivo . However, in contrast to Sei et al . , this group indicated that docetaxel enhances microtubule stabilization, which in turn facilitates reoviral replication and enhances the therapeutic potential of the combination therapy. Further studies on paclitaxel and reovirus therapy found that the proviral state could be induced in endothelial cells exposed to vascular endothelial growth factor (VEGF) during rebound from paclitaxel-mediated inhibition of VEGF signaling and suggest that careful scheduling of combination viro- and chemotherapeutics is necessary when considering future protocols . It is interesting to note that other non-taxane drug combinations such as vincristine and reovirus also resulted in synergistic kill without microtubule stabilization, suggesting evidence of multiple mechanisms by which reovirus may combine with anticancer treatments to enhance its antitumor effects .
Work on multiple myeloma (MM) showed that reovirus enhanced the anticancer activity of bortezomib; however, this finding was not confirmed by isobolographic analysis . The anticancer activity of reovirus and bortezomib was associated with increased levels of NOXA and ER stress, leading to apoptosis in MM . Importantly, no such expression of stress genes is seen in normal peripheral blood mononuclear cells under the same treatment regime. Reovirus also significantly enhanced bortezomib’s activity in an MM xenograft model and a bone disease syngeneic mouse model.
Recently, it was demonstrated that the combination of reovirus with the mTOR inhibitor rapamycin was synergistic in a mouse melanoma model. Although this appears to be sequence dependent in vitro , virus with concomitant administration of rapamycin significantly improves survival in vivo (C. Comins, manuscript in preparation). The mechanism for this synergy is uncertain, but the synergy seen in other oncolytic virus models may be due to a reduction in the neutralizing anti-reoviral antibody (NARA) response caused by rapamycin (C. Comins, manuscript in preparation). However, no significant attenuation in the NARA response was found, which may reflect differences in the dose scheduling of rapamycin or the timing of NARA assessment after viral administration (C. Comins, manuscript in preparation). Whereas viral replication in tumors of treated mice showed a significant increase in the group receiving sequential reovirus and rapamycin compared to the reovirus-only group, there was no significant difference between the sequence or concomitant groups and neither was there a difference between the concomitant and reovirus-only groups. The combination of mTOR inhibitors with reovirus therefore warrants further investigation.
Reovirus Therapy: Clinical Data
Many clinical trials involving reovirus type 3 Dearing (Reolysin, Oncolytics Biotech) have been completed or are ongoing. These include studies assessing different strategies for the delivery of reovirus to the tumor as a single agent, as well as the potential of reovirus in combination with other treatment modalities. A summary of these clinical trials is provided in Table 13.1 .
| Study No. | Phase | Details | Tumor Type | Investigational Site | Status | Outcome |
|---|---|---|---|---|---|---|
| REO1 | I | Single agent, intralesional injection | Advanced solid tumors (pericutaneous) | Tom Baker Cancer Centre, Calgary, Alberta, Canada | Completed 19 patients | No MTD reached |
| REO2 | I | Single agent, intraprostatic injection | Prostate cancer | Tom Baker Cancer Centre, Calgary, Alberta, Canada | Completed 6 patients | No MTD reached |
| REO3 | I | Single agent, intralesional injection | Recurrent gliomas | Tom Baker Cancer Centre, Calgary, Alberta, Canada | Completed 12 patients | No MTD reached |
| REO4 | I | Single agent, intravenous administration | Advanced solid tumors | Montefiore Medical Center, Bronx, NY | Completed 18 patients | No MTD reached |
| REO5 | I | Single agent, intravenous administration | Advanced solid tumors |
| Completed 33 patients | No MTD reached |
| REO6 | Ia | Intralesional administration combined with radiotherapy | Advanced solid tumors (pericutaneous) | Royal Marsden Hospital, Sutton, UK St. James Hospital, Leeds, UK | Completed 12 patients | No MTD reached |
| Ib | ||||||
| Advanced solid tumors (pericutaneous) | Royal Marsden Hospital, Sutton, UK St. James Hospital, Leeds, UK | Completed 11 patients | No MTD reached | |||
| REO7 | I/II | Single agent, intralesional infusion | Recurrent gliomas | University of Alabama, Birmingham, AL | Completed 15 patients | No MTD reached |
| REO8 | II | Intralesional administration combined with radiotherapy | Advanced solid tumors (pericutaneous) |
| Completed 16 patients | No MTD reached |
| REO9 | I | Intravenous administration with concurrent gemcitabine | Advanced solid tumors | Royal Marsden Hospital, Sutton, UK | Completed 16 patients | No MTD reached |
| REO10 | I | Intravenous administration with concurrent docetaxel | Advanced solid tumors |
| Completed 24 patients | No MTD reached |
| REO11 | I | Intravenous administration with concurrent paclitaxel and carboplatin | Head and neck tumors | Royal Marsden Hospital, Sutton, UK | Completed 31 patients | No MTD reached |
| REO12 | I | Intravenous administration combined with cyclophosphamide | Advanced solid tumors |
| Commenced March 2008 | No MTD to date |
| REO14 | II | Single agent, intravenous administration | Metastatic sarcomas (bone and soft tissue) | The University of Texas Health Science Center at Houston, Houston, TX | Completed 53 patients | – |
| NCI trial | II | Intravenous and Intraperitoneal administration | Metastatic ovarian tumors | National Cancer Institute, Bethesda, MD | Commenced mid-2008 | – |
| REO15 | II | Intravenous administration in combination with paclitaxel and carboplatin | Head and neck tumors | The University of Texas Health Science Center at Houston, Houston, TX | Ongoing | – |
| REO16 | II | Intravenous administration in combination with paclitaxel and carboplatin | Non-small-cell lung cancers | The Ohio State University Comprehensive Cancer Center, Columbus, OH | Ongoing | – |
| REO17 | II | Intravenous administration in combination with Gemcitabine | Advanced pancreatic cancer | The University of Texas Health Science Center at Houston, Houston, TX | Ongoing | – |
| REO18 | III | Intravenous administration in combination with paclitaxel and carboplatin | Squamous cell carcinomas of the head and neck | International | Ongoing | – |
| OSU-07022 (NCI Trial) | I/II | Systemic and intraperitoneal administration | Metastatic ovarian, peritoneal, and fallopian tube cancers | National Cancer Institute, Bethesda, MD | Ongoing | – |
| MAYO-MC0672 (NCI trial) | II | Intravenous administration | Metastatic melanoma | National Cancer Institute, Bethesda, MD | Complete | – |
| REO20 | II | Intravenous administration combination with paclitaxel and carboplatin | Metastatic melanoma | The University of Texas Health Science Center at Houston, Houston, TX | Ongoing | – |
| REO21 | II | Intravenous administration combination with paclitaxel and carboplatin | Squamous cell carcinoma lung cancer | The University of Texas Health Science Center at Houston, Houston, TX | Ongoing | – |
| REO22 | I | Intravenous administration combination with FOLFIRI | Colorectal cancer | Montefiore Medical Center at the Albert Einstein College of Medicine, New York | Ongoing | – |
| GOG-0186H (NCI/GOG trial) | II | Intravenous administration of combination with paclitaxel | Persistent or recurrent ovarian, fallopian tube, or primary peritoneal cancer | National Cancer Institute, Bethesda, MD | Ongoing | – |
| COG-ADVL1014 (NCI/COG trial) | I | Intravenous administration combination with cyclophosphamide | Pediatric patients with relapsed or refractory solid tumors | National Cancer Institute, Bethesda, MD | Ongoing | – |
| OSU-10045 (NCI trial) | II | Intravenous administration combination with paclitaxel and carboplatin | Metastatic pancreatic cancer | National Cancer Institute, Bethesda, MD | Ongoing | – |
| OSU-11148 (NCI trial) | I | Intravenous administration of Reolysin | Relapsed multiple myeloma | National Cancer Institute, Bethesda, MD | Ongoing | – |
| IND 209 (NCIC CTG trial) | II | Intravenous administration combination with docetaxel | Metastatic castration-resistant prostate cancer | Canada | Announced | – |
| IND 210 (NCIC CTG trial) | II | Intravenous administration combination with FOLFOX-6 plus bevacizumab (Avastin) versus FOLFOX-6 | Advanced or metastatic colorectal cancer | Queen’s University, Kingston, Ontario, Canada | Announced | – |
| IND 211 (NCIC CTG trial) | II | Intravenous administration combination with docetaxel or pemetrexed | Advanced or metastatic non-small-cell lung cancer | Queen’s University, Kingston, Ontario Canada | Announced | – |
| IND 213 (NCIC CTG trial) | Phase II | Intravenous administration combination with paclitaxel | Patients with advanced or metastatic breast cancer | Queen’s University, Kingston, Ontario Canada | Announced | – |
Related posts:
 Modified Oncolytic Herpesviruses for Gene Therapy of Cancer
Clinical Application of Plasmid-Based Cancer Vaccines
Modified Oncolytic Herpesviruses for Gene Therapy of Cancer
Clinical Application of Plasmid-Based Cancer Vaccines
 Imaging of Oncolytic Virus Gene Expression
Ethics in Translational Gene Transfer Research
Imaging of Oncolytic Virus Gene Expression
Ethics in Translational Gene Transfer Research
 Viral Insertion Site Detection and Analysis in Cancer Gene Therapy
Viral Insertion Site Detection and Analysis in Cancer Gene Therapy
 Selectively Replicating Oncolytic Adenoviruses Combined with Chemotherapy, Radiotherapy, or Molecular Targeted Therapy for Treatment of Human Cancers
Selectively Replicating Oncolytic Adenoviruses Combined with Chemotherapy, Radiotherapy, or Molecular Targeted Therapy for Treatment of Human Cancers
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


