The average 70-kg person has 42 L of water divided into the intracellular space (ICF) of 28 L and the extracellular space (ECF) of 14 L. The ICF is subdivided into the red blood cell (RBC) mass of 2 L and the visceral mass of 26 L; the ECF is subdivided into a plasma volume (PV) of 3 L and an interstitial fluid space (IFS) volume of 11 L. The cardiac output (CO) in this 70-kg person is 5 L/min with 20% of this flow going to kidneys; thus the kidneys, with a combined weight of about 600 g, have a renal blood flow (RBF) of 1250 mL/min or more than 2 mL/min/g of renal parenchymal. This unusually large RBF reflects the vital renal role in regulating the ICF and ECF, controlling fluid and electrolyte balance, modulating acid-base balance, and excreting undesirable catabolyes.1,2 Protection of renal function is essential for recovery after a shock or septic insult. This chapter reviews normal renal physiology, the renal response to shock and sepsis, guidelines for prevention and treatment of acute renal failure (ARF).
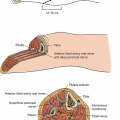
The 1250 mL/min of RBF passes through the renal artery into the interlobar, the arcuate, and, finally, the intralobar arteries; 85% of the RBF perfuses the outer cortical glomeruli; the remaining 15% of RBF perfuses the juxtamedullary glomeruli (Fig. 59-1). The glomeruli (Bowman’s capsules) are like capillaries except that proteins, normally, are not filtered.2,3 While passing through the glomeruli, 20% of the plasma is filtered as a cell-free, protein-free filtrate. The effective renal plasma flow (ERPF) through these tubular vessels is determined by the clearance of para-aminohippurate (CPAH) that is filtered and secreted but not reabsorbed by the renal tubules. Ninety-one percent of PAH is cleared in one passage; 9% remains bound to the plasma protein. Renal oxygen consumption parallels ERPF which averages 650 mL/min. True renal plasma flow (TRPF) is calculated by dividing ERPF by 0.91 and averages 710 mL/min. The extraction ratio of PAH (EPAH), however, may vary with injury and sepsis; true EPAH requires renal vein sampling to accurately measure true renal plasma flow (TRPF) (TRPF = ERPF/EPAH).2,3,4,5 TRBP may be calculated by correcting the TRPF for hematocrit: TRBF = TRPF/(1-Hct).
FIGURE 59-1
The kidney is divided into three components. The outer cortex (CI) contains 85% of the glomeruli. The inner cortex/outer medulla (CII) contains the remaining juxtamedullary glomeruli whose peritubular vessels extend to the vasa recta in the inner medulla (CIII) that establishes the hyperosmolality within the loops of Henle.
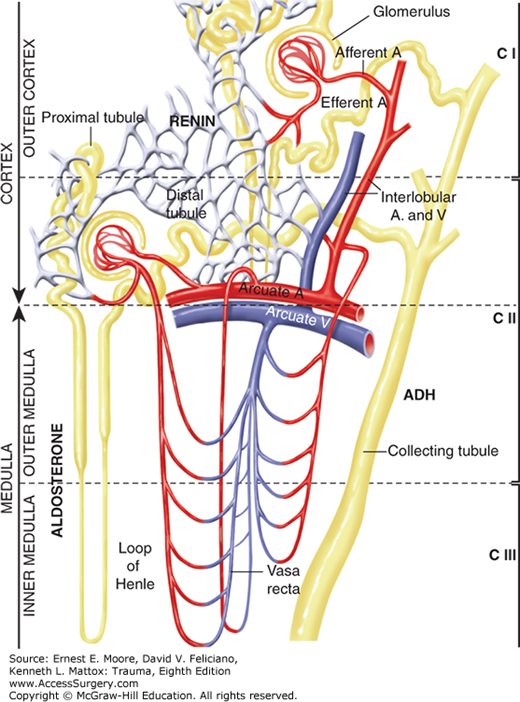
The distribution of RBF can be measured by isotopic disappearance of radioactive xenon-133 (133Xe) or krypton-85 that can be graphically portrayed as a cumulative slope composed of four separate subslopes reflecting parallel flow in mL/min per 100 g to components CI (outer cortex), CII (inner cortex and outer medulla), CIII (inner medulla), and CIV (renal pelvis and fat) (Fig. 59-1).2,5,6 Blood leaving the juxtamedullary component II glomeruli perfuses the peritubular vessels to the long straight vasa recta in the inner medulla prior to returning to the small veins adjacent to the same glomerulus.2,3
The normal glomerular filtration rate (GFR) averages 125 mL/min (180 L/d) and is measured by the renal clearance (urine concentration × volume/plasma concentration) of exogenous inulin (Cin) or endogenous creatinine (Ccr), both of which are completely filtered; the Cin is slightly higher than the Ccr since creatinine in humans is partially reabsorbed by the renal tubules.3,7 The work of GFR is performed by the heart. The protein-free filtrate passes through the proximal convoluting tubules where approximately 80% of the sodium and water are reabsorbed by active sodium transport and the passive movement of water, thereby, maintaining an isomolar state. The nonfiltered blood, now protein-rich, perfuses the efferent arterioles and the peritubular vessels, and augments tubular reabsorption and secretion.8
The juxtaglomerular (CII) nephrons that receive 15% of the RBF are unique; each has a loop of Henle with descending and ascending straight segments that pass into the inner medulla.2,3 These segments actively reabsorb sodium against a gradient and, thereby, create a hypertonic medullary interstitium (CIII) that facilitates the subsequent concentration of glomerular filtrate and the preservation of salt and water in response to antidiuretic hormone and aldosterone. Glucose and electrolytes, namely, chloride, phosphate, and potassium, are likewise absorbed at this site. This hypertonic medullary interstitium is further regulated by the peritubular vessels and vasa recta passing close to the loop of Henle (Fig. 59-1). Rapid blood flow through these vessels may cause a “washout” of sodium ions and osmoles resulting in transient “paralysis” of the renal concentrating mechanism; inadequate blood flow with ischemia injury to these tubular cells impedes sodium reabsorption against a gradient, thus preventing medullary hypertonicity and normal filtrate preservation.2,3 This is discussed later.
Twenty percent (ie, 36 L/d) of protein-free filtrate and sodium enters the distal convoluting tubules where additional sodium is reabsorbed by active transport as chloride follows passively.3,8 This process is facilitated by aldosterone. The distal tubular aldosterone effect is estimated by dividing the free water clearance (CH2O) by the CH2O + sodium clearance (CNa) in patients with a positive CH2O. When the product is greater than 0.73, the “aldosterone” effect has been blocked. The distal tubule also exchanges sodium for either potassium or hydrogen depending on pH and potassium load. Each distal tubule returns to its own glomerulus at the afferent arteriole where the macula densa and Polkissen body, known as the juxtaglomerular apparatus (JGA), are located. The JGA serves as a “feedback” loop for each nephron affecting sodium and water balance which are mediated by distal tubular sodium concentration, afferent arteriole pressure, afferent arteriole pulse pressure, and others.3,7,8,9
After passing through the distal convoluted tubules, the remaining hypotonic filtrate (18 mL/min or 36 L/d) enters the collecting ducts where water reabsorption occurs; this is facilitated by antidiuretic hormone (ADH) as the filtrate passes through the hypertonic inner medullary (CIII) interstitium to the renal pelvis.3 The final concentration and urine volume, therefore, vary with PV, serum osmolality, ADH release, and other factors; urine concentration may range from isomolar to 1400 mOs/L. Alteration of this highly integrated system occurs with hemorrhagic shock so that the renal concentrating ability may be restricted to 600 mOs/L.4 The collecting ducts normally reabsorb approximately 17 mL/min or 34 L/d with the remaining 1 mL/min (1440 mL/d) being excreted as urine.
The renal handling of sodium, osmoles, and water is expressed as the clearances of sodium (CNa), osmoles (Cosm), and free water (UV – Cosm).4 With normal water and solute intake, the CNa and Cosm average 1–3% of the GFR; CH2O is usually negative, reflecting the excretion of concentrated urine. A decrease in RBF or PV causes CNa and Cosm to fall due to sodium preservation.3 Likewise, a breakdown in the counter current mechanism brought about by a selective insult to the juxtamedullary nephrons and their loops of Henle causes increased CNa and Cosm from impaired tubular concentration of sodium.3,4
The very high ratio of RBF to kidney weight allows the kidney to efficiently preserve PV after injury and hemorrhage (Table 59-1).3,10 Renal vasoconstriction at the efferent arteriole permits a rise in renal vascular resistance (RVR) from a normal 5000–8000 dyne s/cm5 with a concomitant decrease in RBF from 1250 to 800 mL/min while maintaining a normal GFR (Fig. 59-2). This phenomenon of maintained GFR despite a reduction in RBF is known as autoregulation (Fig. 59-2). Excretion of metabolites is thus maintained, while 400 mL of blood/min is redirected to core areas. Both experimental and clinical studies show that autoregulation allows GFR to be maintained while RBF is decreased to 70% of normal.2,3,11 The filtration fraction (GFR/ERPF) under such circumstances increases from a normal of 20% to as high as 40%.3,11 More severe hypovolemia causes vasoconstriction at both the afferent and efferent arterioles, thus leading to a reduction in GFR (Table 59-1). The mechanism for the rise in RVR is multifactorial due primarily to the renal perfusion of peripherally generated catecholamines that, in turn, activates the JGA to stimulate intrarenal renin release. This stimulates the renin-angiotensin-aldosterone system (RAAS) which not only promotes sodium reabsorption but may also increase RVR.7,12,13 When the RVR increases above 14,000 dyne s/cm5, the RBF falls below 500 mL/min, thereby allowing over 700 mL/min to be redirected to core organs. When hypovolemia causes hypotension below 70 mm Hg, GFR ceases and essentially all RBF is redirected to the systemic circuit (Table 59-1). This causes renal ischemia and potentiates ARF.
| Severity of hemorrhage | Volume deficit (mL) | Blood pressure (torr) | Renal plasma flow (mL/min) | Filtration rate (mL/min) | Sodium clearance (mL/min) | Osmolar clearance (mL/min) | Urine output (mL/min) | Renal vascular resistance (dyne s/cm5) |
|---|---|---|---|---|---|---|---|---|
| Normal | 5000 | 120/80 | 650 | 125 | 1.5 | 2.0 | 1.0 | 6000 |
| Class I | 700 | 120/80 | 520 | 115 | 1.3 | 2.0 | 0.9 | 5500 |
| Class II | 1200 | 115/80 | 260 | 60 | 0.7 | 1.3 | 0.3 | 8000 |
| Class III | 1600 | 90/70 | 85 | 10 | 0.1 | 0.1 | 0.1 | 10,000 |
| Class IV | 2000 | 60/40 | 10 | 2 | 0 | 0 | 0 | 12,000 |
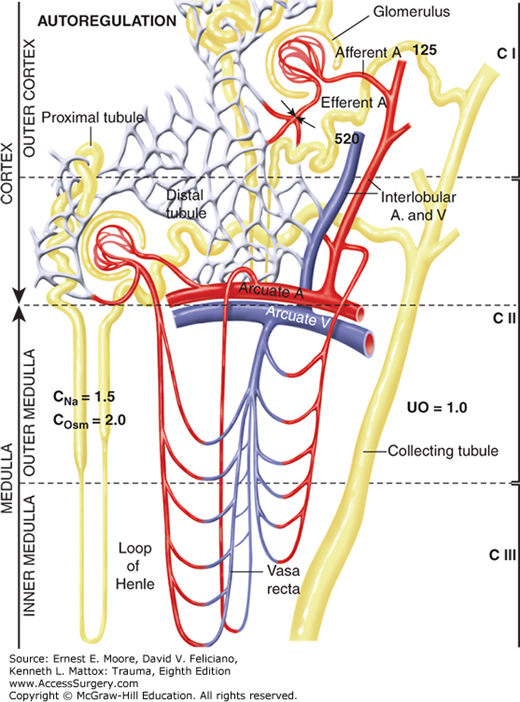
During hypoperfusion, the kidney conserves salt and water due to decreased GFR, increased ADH, and increased renin release with aldosterone generation.3,13 When PV and CO are restored, the renal vasoconstriction subsides—first at the preglomerular afferent arteriole and later at the postglomerular efferent arteriole. The increase in RVR, however, may persist for many hours and even days in patients with a severe hemorrhagic shock.3,13,19
During operation, the kidney exerts the same autoregulatory response to a PV deficit as described above. The major difference reflects the altered humoral response brought about by general anesthesia, especially in the marginally volemic patient in whom systemic vasoconstriction was maintaining a blood pressure prior to induction. This was first described by Ladd during the Korean conflict.12 Civilian studies have shown the same phenomenon.2,3 The sudden reduction in protective vasoconstriction plus continued bleeding from injured organs precipitates a marked reduction in PV and CO causing hypotension, increased RVR, and decreased RBF with an abrupt reduction in CI flow; the consequent fall in GFR causes oliguria or anuria, which may persist as ARF after operation.2,12
The prime objective during operation is to correct the depleted PV and ineffective CO while hemostasis is obtained. When hemostasis has been achieved and the blood pressure has been restored, oliguria often persists. Osmotic or loop diuretics, such as mannitol or furosemide, have been advocated in this setting on the assumption that induced diuresis increases RBF, GFR, and urine output, and prevents ARF.2,14 Most studies, however, show that diuresis in this setting provides no renal protection.16,17 Loop diuresis in combination with low-dose dopamine, likewise, affords no renal protection during major surgery.17 Induced diuresis causes a further decrease in effective PV, thus making the likelihood of ARF greater.18 Furthermore, induced diuresis interferes with one of the crucial monitors of effective postoperative PV replacement, namely, uninduced urine output rates.
Intraoperative protection of the kidney in a hypovolemic, hypotensive patient must be directed toward obtaining hemostasis and improving the cardiovascular status by PV expansion with crystalloid solution, blood, and blood products. Inotropic support should be added when PV expansion causes an elevated central pressure despite persistent hypotension and oliguria. Resuscitation in his setting is best accomplished by a balanced electrolyte solution rather than 0.9% saline which may contribute to ARF by the resultant hyperchloremia and acidosis.18 Experimental and clinical studies show that a temporary delay in reestablishing urine flow after mean arterial pressure (MAP) is restored results from a prolonged rise in RVR after hypovolemia is corrected.7,10 Renal vasodilation, experimentally, can reverse this lagging oliguria, but such therapy is difficult and hazardous in humans.10 Based on clinical observations, many patients in whom the MAP has been restored are still PV and IFS depleted, resulting in a marked increase in both total peripheral resistance (TPR) and RVR. The persistent elevation in RVR is likely due to the prior ischemic insult and not perfusion by catecholamines in stable patients; measurements of renin and arginine vasopressin (AVP) at this time have been normal (C. E. Lucas and I. K. Rosenberg, Surgical renal study unit, unpublished data). Thus, pharmacologic intervention with angiotensin-converting enzyme blockade or nitrous oxide would likely be ineffective.13 Since both peripheral and renal vasoconstriction can lead to a doubling of total resistance, the CO may be reduced by 50% of normal despite a normal MAP. Thus, the PV may be only 1500 mL after the MAP rises to normal. Assuming that concomitant oliguria reflects this vasoconstriction and PV depletion, the rapid infusion of 1 or 2 L of balanced electrolyte solution plus whole blood, if indicated, will restore RBF, GFR, and urine output. This approach protects renal function in the early postoperative period. The few patients who are unresponsive to this regimen may be treated by a loop diuretic, such as furosemide (40 mg), which typically produces a dramatic diuresis. Such patients, however, must be monitored closely since even small dosages of a loop diuretic may induce excessive diuresis, leading to subsequent hypovolemia and hypotension.16,17
An interesting, but potentially hazardous, renal response to shock in injured patients is a transient period of polyuria during or immediately after operation (Fig. 59-3).19 This polyuria is not excessive, seldom exceeding 250 mL/30 min, and usually abates by 5 hours following operation (Table 59-2). This phenomenon tends to occur in patients who have had a major hemorrhagic shock insult requiring more than 15 blood transfusions prior to successful hemostasis.19 On arrival to the SICU, the blood pressure and pulse normalize while the urine output exceeds 3 mL/min (ie, 180 mL/h) at the expense of effective PV. The urine sodium concentration exceeds 40 mEq/L and the fractional excretion of sodium (FENa) or CNa exceeds 3%. Therapy for this syndrome should be by maintenance of effective PV as judged by vital signs until the polyuric phase subsides, usually by 2–5 hours after operation.19
FIGURE 59-3
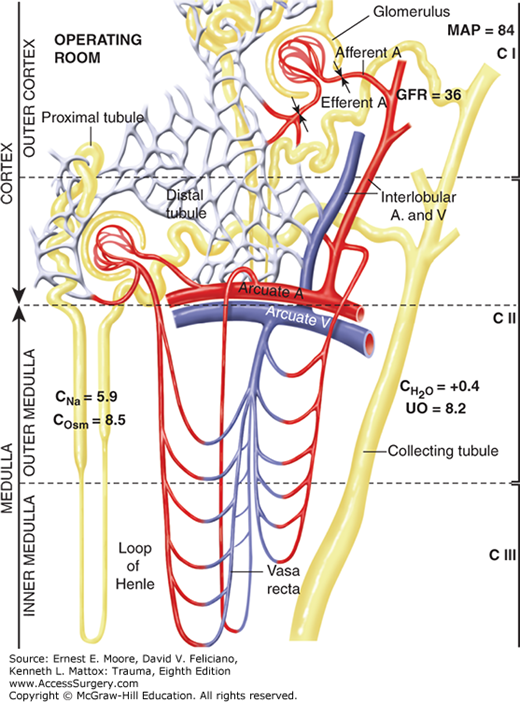
Following restoration of PV during and shortly following operation, a renal concentrating defect may exist whereby a patient may have a low-normal MAP (84 torr) with a markedly reduced GFR (35 mL/min) while excreting a large urine volume (8.2 mL/min; 500 mL/h). This concentrating defect is likely due to inner medullary washout of osmoles, thus impairing reabsorption of sodium and water in response to aldosterone and AVH. This phenomenon seldom lasts more than 5 hours following conclusion of operation.
The mechanism for early postoperative polyuria is unclear but likely is due to an inner medullary (CIII) washout of osmoles. During shock, the primary reduction in RBF occurs in the outer cortex (CI) with less reduction in juxtamedullary (CII) flow; this is referred to as cortical to medullary shunting.2,3 Since the juxtamedullary nephrons have loops of Henle that affect interstitial medullary (CIII) tonicity, a relative increase in RBF to these nephrons may cause a “washout” of osmoles. This disrupts the countercurrent regulatory system and precludes effective sodium and water reabsorption from the collecting ducts when outer cortical flow is first reestablished.19
Increased osmotic diuresis with polyuria may also be due to the overutilization of solutions containing 5% dextrose. Marked hyperglycemia (500–1000 mg/100 mL) and hyperosmolemia (310–320 mOs/L) have occurred in patients resuscitated exclusively with crystalloid solutions containing 5% dextrose. This hazard is circumvented by limiting the amount of 5% dextrose in crystalloid solution to 2000 mL after which a nonglucose balanced electrolyte solution is infused along with whole blood and blood products as needed.2,19
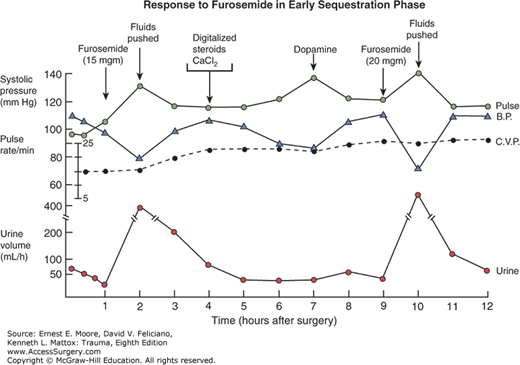
Following operative control of bleeding after severe hemorrhagic shock, there are major shifts in sodium, water, and protein from the plasma into the IFS. This causes reduced PV, MAP, CO, RBF, GFR, and urine output. This phase of extravascular fluid sequestration lasts about 36 hours in patients who have received an average of 15 RBC transfusions prior to operative control of bleeding.20 Part of the IFS expansion includes intrapulmonary sequestration resulting in respiratory insufficiency.20 Successful therapy mandates a careful balance to maintain perfusion and protect the kidney while not overloading the pulmonary circulation (Fig. 59-4).
FIGURE 59-4
Following an operation requiring 23 RBC units and 9 L crystalloid solution, this patient became oliguric (1 hour) and was treated with low-dose furosemide causing prompt diuresis and hypotension. Fluid bolus restored pressure (2 hours) and inotropes were added for rising central pressures (steroid use was part of a prospective randomized trial). Low-dose dopamine was added at 7 hours and this improved perfusion pressure. Furosemide at hour 9 reproduced prompt diuresis and hypotension treated with additional fluid bolus through hour 10. Clearly, loop diuresis during the fluid update phase is hazardous. The patient made a full recovery.
When the intravenous infusion rate is decreased because of increased weight gain and IFS sequestration, the MAP falls, causing a fall in RBF and potential ARF. Mesenteric leukotrienes which colocalize with 5-lipoxygenase activating protein and may contribute to ARF after hemorrhagic shock; mesenteric lymph diversion of the 5-lipoxygenase ameliorates the subsequent kidney insult in a rodent hemorrhagic shock model.21 This typically occurs during the initial 12 hours after operation and causes decreased ERPF, GFR, UO, CNa, and COsm. During this period of obligatory IFS expansion, the patient may need many liters of balanced electrolyte solution to maintain kidney perfusion. Inotropes and ventilator support are often needed.21 Restoring PV, hemoglobin level, coagulation status, and normal MAP enables the kidney to maintain GFR and urine output even though RBF may be reduced. CNa and COsm during this period reflect the underlying renal circulatory status and are decreased when RBF is lowered but return to normal when the effective circulatory volume is restored; CH2O is negative at this time.11 Excess crystalloid resuscitation, especially, during the latter portion of the fluid uptake phase is detrimental causing acute lung injury, cardiac compromise, and, even, the abdominal compartment syndrome. Restricting crystalloids, however, will not prevent IFS expansion but will lead to PV depletion with reduced perfusion with the need for inotropic support and vasopressors. This sequence promotes ARF which, in the fluid sequestration phase, usually results in death.2,20,22 A therapeutic balance is required.
The technique used to provide ventilatory support during the fluid uptake phase may also affect kidney function.23,24 High tidal volumes (>10 mL/kg body weight), with or without high positive end-expiratory pressure (PEEP > 10 cm H2O) may improve oxygen tension by reducing atelectasis but compromise oxygen delivery reducing CO.23 This reduces MAP, CO, ERPF, CNa, COsm, and urine output.24 The renal response is mediated through sinoaortic baroreceptors and renal vein pressure.25,26 The SICU team must adjust ventilatory settings with renal function in mind.
Colloid supplementation during the sequestration phase also affects renal function.27 Human serum albumin (HSA) therapy given to prevent weight gain and IFS expansion during the fluid uptake phase, ignores the fact that 7% of serum albumin exits the PV into the IFS each hour and increases when supplemental HSA is administered; this causes a prolonged fluid uptake phase, greater crystalloid needs, greater weight gain, and compromised renal dynamics.27,28 Patients receiving HSA during the sequestration phase exhibit an increase in PV and RBF but, paradoxically, a reduction in GFR, CNa, Cosm, and urine output.28 The reduction in GFR is caused by the HSA oncotic properties within the glomerular tufts where Bowman’s capsule acts like a modified capillary except that it is impervious to protein transmigration. The hyperoncotic blood leaving the glomerular tufts perfuses the vasa recta in the inner medulla (CIII) and extracts excess water and sodium from the interstices of the inner medulla. The resultant hyperosmolar CIII interstices facilitate the reabsorption of sodium and water from the distal nephrons under the influence of these facultative hormones (Table 59-3).2,3,21 Patients receiving HSA supplementation require more frequent loop diuresis and are more likely to develop ARF compared with patients not receiving supplemented HSA.21,28 This phenomenon has been duplicated in a canine model of isolated renal perfusion (C. E. Lucas and I. K. Rosenberg, Surgical renal study unit, unpublished data).21
The hypotension associated with IFS expansion after operation often stimulates therapy with vasopressor agents to restore MAP by precapillary vasoconstriction. The resultant rise in MAP may stimulate the baroreceptor response so that the pituitary reduces the release of ADH, thereby, promoting increased urine output. This increase in urine volume can be duplicated initially with all vasopressor agents when administered at a low dosage.29 Unfortunately, when tachyphylaxis to the vasopressor agent occurs, increased vasopressor doses must be administered; the resultant vasoconstriction affects the renal circulation and potentiates ARF.
Related posts:



Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


