Fig. 6.1
Regulatory T cells (Tregs) limit self-directed immune responses but also suppress antitumor immunity and clearance of chronic viral infections. Tregs are essential for the maintenance of peripheral tolerance and control immune homeostasis through suppression of autoimmunity, limiting allergic immune responses and preventing damaging immune responses to pathogens. However, because of these normal immunosuppressive functions, Tregs also have negative consequences in that they can prevent effective antitumor immunity and limit clearance of chronic viral infections
6.2 Discovery of a T Cell Population that Regulates Autoimmunity
Tregs were first discovered as a subpopulation of CD4+ T cells that were responsible for preventing autoimmunity. Subsequent work over several decades has elucidated molecular pathways and surface receptors associated with Tregs and has led to an appreciation for the central role they play in maintaining peripheral tolerance in vivo. As such, research into the role of Tregs in cancer continues to expand, especially in the context of immunotherapeutic targeting. An initial discussion of the discovery and elucidation of surface markers for the identification of this T cell population is warranted to establish a basic understanding of the role of Tregs in immune homeostasis.
6.2.1 Suppression of Autoimmunity by CD4+ T Cells
The importance of Tregs was first described in studies of autoimmunity in animal model systems dating back to the mid-1960s. These early studies showed that removal of the thymus from neonatal mice led to severe autoimmunity in many organs, including hematological disorders, endocrinopathies, gastritis, and oophoritis/orchitis [3]. These studies demonstrated an important role for T cells derived from the thymus in suppressing immune responses in a variety of tissues. Importantly, these early studies also demonstrated that a T cell subpopulation was required to prevent autoimmunity as the transfer of CD4+CD8−, but not CD4−CD8+, thymocytes was sufficient to abrogate autoimmunity (Fig. 6.2a).
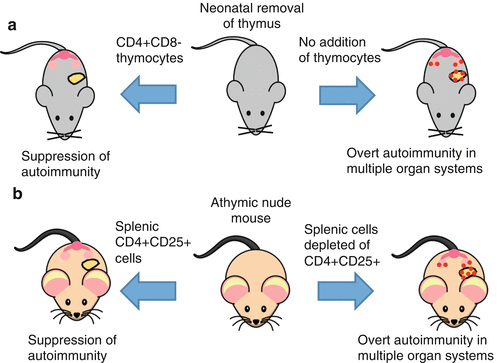
Fig. 6.2
Discovery of a T cell population that suppresses autoimmunity. (a) When the thymus is removed from neonatal mice within 3 days of birth, overt autoimmunity occurs in diverse organ systems such as the ovaries and pancreas. However, this autoimmunity can be prevented when CD4+CD8− thymocytes are used to reconstitute these athymic mice. (b) Subsequent work showed that autoimmunity occurred in multiple organ systems when naturally athymic nude mice were reconstituted with splenic cells depleted of CD4+CD25+ T cells. Conversely, reconstitution of nude mice with splenic CD4+CD25+ T cells led to the suppression of autoimmunity. Collectively, these experiments demonstrated that thymus-derived CD4+CD25+ T cells that develop soon after birth are responsible for the suppression of autoimmunity
6.2.2 Markers of Regulatory T Cells
Subsequent studies sought to define cell-intrinsic markers of this suppressive cell population. Studies to identify markers of Tregs first focused on the observation that Tregs appeared to be an activated T cell population. A cell surface marker that is associated with T cell activation and function is the high-affinity IL-2 receptor α-chain (IL2Rα or CD25). The importance of CD25 in Treg biology was discovered by comparing reconstitution of athymic nude mice with CD4+CD25+ versus CD4+CD25− splenocytes. Reconstitution of nude mice with the CD4+CD25+ subpopulation led to suppression of autoimmunity, while the CD4+CD25− T cell-reconstituted mice succumbed to autoimmunity (Fig. 6.2b) [4]. This study was the first to highlight the importance of CD25 as a marker of Tregs.
Advancing these observations demonstrated that both CD25 and IL-2 are essential for Treg development and survival [5]. Although IL-2 contributes to survival and function of all T cells via CD25, it is critically important for Tregs, so much so that CD4+CD25+ T cells are highly enriched for Tregs. Importantly, Tregs do not secrete IL-2 themselves and therefore require paracrine production of IL-2 by other cell types to exert their effector function. Following the identification of CD4+CD25+ T cells as a suppressive cell population, additional studies sought to further define markers of Tregs.
The discovery of a genetic mutation that led to a severe autoimmune disease in mice (known as scurfy), and a similar disease in humans (known as immunodysregulation polyendocrinopathy enteropathy X-linked syndrome or IPEX), led investigators to consider the role of this gene in Tregs. This single X-linked gene (now known as Foxp3) was found to encode a key transcription factor (FOXP3) that directs the formation and function of Tregs [6]. FOXP3 has been shown to be critical in driving the development and suppressive function of Tregs by controlling the transcription of key genes required for the maintenance and function of Tregs. FOXP3 drives or enhances the transcription of genes associated with suppression, such as CD25 and CTLA-4, while simultaneously suppressing the transcription of inflammatory genes, such as IFN-γ and IL-2. Expression of FOXP3 itself is driven by the epigenetic hypomethylation of the Foxp3 promoter, which is considered a hallmark of Tregs [7]. While FOXP3 is a specific marker for Tregs, it should be noted that FOXP3 can also be transiently expressed in activated human CD4+ T cells [8]. This transient expression of FOXP3 following activation can cause some effector T cells to appear to be FOXP3+ Tregs, and therefore phenotypic identification of human Tregs should rely on a combination of markers (i.e., coexpression of CD25 and FOXP3 or the absence of CD127, as described below). Additionally, FOXP3 cannot be used to sort Tregs from unmanipulated samples from mouse or human donors, as it is a transcription factor that is expressed in the nucleus. Consequently, murine studies in which FOXP3+ cells are purified routinely rely on the use of genetic reporters.
More recently, the absence of the interleukin-7 (IL-7) receptor (CD127) on CD4+CD25+ T cells has been described as a population that is enriched for FOXP3+ Tregs. Conventional memory T cells require signals from IL-7 for their maintenance and as such express high levels of CD127 [9]. Conversely, Tregs do not express CD127 because FOXP3 suppresses transcription of the Il7r gene, leading to the absence of CD127 on cells that express FOXP3 [10]. In addition to being enriched for expression of FOXP3, cells that are CD4+CD25+CD127− are highly suppressive in vitro, demonstrating that this population is functionally Tregs. Collectively, these markers have facilitated the purification and analysis of Tregs. However, identification of additional markers would be beneficial.
6.2.3 Regulatory T Cell Origins: Thymus Versus Periphery
The role of Tregs in preventing autoimmunity was described using thymus-derived Tregs (tTregs). However, populations of suppressive CD4+FOXP3+ T cells can also be generated outside of the thymus [11]. Tregs that develop in vivo outside of the thymus are known as peripheral Tregs (pTregs). pTregs differentiate from naïve CD4+ T cells in the periphery following activation of naïve CD4+ T cells with suboptimal doses of antigen in the presence of transforming growth factor β (TGF-β). Activation of naïve CD4+ T cells under these conditions leads to the induction of FOXP3, the inability to secrete the effector cytokines IFN-γ and IL-2, and the ability to suppress proliferation of effector T cells in vitro. While tTregs clearly limit autoimmunity in vivo, the role that pTregs play is less clear [12]. One proposed function of pTregs is to suppress immune responses to potentially damaging antigens, such as the gut microbiota, that are not recognized by the self-directed T cell receptor repertoire of tTregs. Alternatively, pTregs may be important for controlling immune responses in specific situations, such as in response to mucosal inflammation [13] or in controlling fetal-maternal tolerance [14]. It is clear that pTregs can be induced in specific situations or to specific antigens in vivo, but their contribution relative to tTregs in controlling autoimmunity and ultimately their role in cancer immunology requires further investigation. Taken together, tTregs are indispensable for limiting autoimmunity in vivo, while pTregs most likely play a role in controlling immune activation in specific scenarios where exogenous antigen-specific Tregs are required.
The identification of a CD4+ T cell population that is essential to the prevention of autoimmunity has led to an entire branch of immunology dedicated to their study. Progress over several decades has led to substantial insight into the role of Tregs in suppressing autoimmunity, the origin and development of Tregs, and the appreciation for the essential role that FOXP3 plays in driving Treg development and function. Identification of cell surface markers for Tregs has also accelerated their analysis in vitro and in vivo.
6.3 Regulatory T Cell Suppressive Mechanisms
Considerable attention has been devoted to understanding the mechanisms by which Tregs suppress immune responses. Broadly speaking, this can be broken down into two classes: mechanisms that are contact dependent and mechanisms that are mediated by soluble factors (Fig. 6.3) [15]. Contact-dependent mechanisms rely on direct interaction of Tregs with the cell types that are being actively suppressed. Soluble suppression mechanisms depend on either Treg secretion of cytokines or metabolic inhibition of effector cells by Tregs. Both types of suppressive mechanisms can also be modulated and potentiated by the local microenvironment and cell-extrinsic pathways, as discussed below.
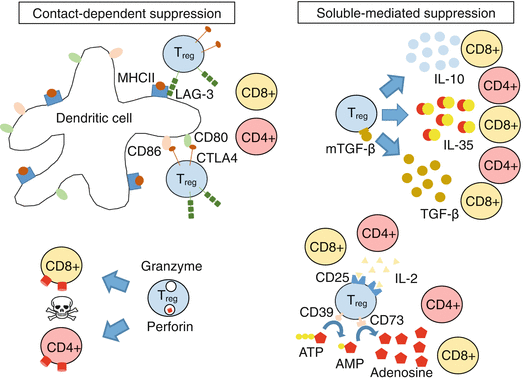
Fig. 6.3
Immunosuppressive mechanisms used by Tregs. Tregs suppress immune responses through either contact-dependent mechanisms or soluble mediators. Contact-dependent inhibition is achieved through interaction of CTLA-4 on Tregs with CD80/CD86 on dendritic cells or through interaction of LAG-3 on Tregs with major histocompatibility complex II on dendritic cells. The interaction of CTLA-4 with CD80/CD86 on dendritic cells prevents co-stimulation of CD28 on effector T cells with CD80/CD86, while LAG-3 prevents TCR/CD3-mediated activation of effector T cells. Expression of granzyme B and perforin in Tregs can lead to the suppression of immune responses through contact-dependent direct cytolysis of effector T cells. Tregs also can suppress effector T cells through soluble cytokines, such as IL-10, IL-35, and TGF-β. Alternatively, Treg suppression with soluble mediators also occurs through metabolic disruption of effector T cells. This occurs through preferential uptake of IL-2 by Tregs due to high expression levels of CD25 (IL2Rα). The presence of the ectoenzymes CD39 and CD73 on the surface of Tregs can catalyze the breakdown of ATP into adenosine, which then can suppress effector T cells or dendritic cells
6.3.1 Contact-Dependent Suppression of Immune Responses
Early studies suggested that Tregs required direct contact with effector T cells or antigen-presenting cells to mediate suppression [16, 17]. Through physical interaction with either conventional CD4+ CD25- T cells or antigen-presenting cells, Tregs limited the production of IL-2 from effector T cells and prevented co-stimulation of effector T cells by antigen-presenting cells. Treg-mediated suppression was lost following the addition of IL-2 or anti-CD28, underscoring that suppression by Tregs relied on deprivation of IL-2 and co-stimulation. These initial studies laid the framework for more in-depth analysis of the contact-dependent mechanisms used by Tregs to suppress immune responses.
One surface molecule expressed by Tregs that mediates contact-dependent suppression is cytotoxic T-lymphocyte-associated protein 4 (CTLA-4). CTLA-4 is required to prevent systemic autoimmunity, and knockout of this gene in mice leads to fatal autoimmune-mediated destruction of multiple tissues [18]. CTLA-4 competes with CD28 for binding to the dendritic cell-expressed co-stimulatory molecules CD80 and CD86. Compared with CD28, CTLA-4 binds to CD80/CD86 with a higher affinity, effectively depriving conventional T cells of co-stimulation. Importantly, CTLA-4 is constituteively expressed by Tregs and mediates one form of contact-dependent suppression. Tregs can also further deprive effector T cells of co-stimulation through trans-endocytosis and subsequent degradation of CD80/CD86 from antigen-presenting cells [19]. In head and neck cancer patients, treatment with targeted chemotherapy led to an increase in intratumoral CTLA-4+ Tregs, which was associated with poor clinical outcome [20]. Contact-dependent immunosuppression by CTLA-4+ Tregs is necessary to maintain immune homeostasis, and CTLA-4+ Tregs are likely to play a role in suppressing antitumor immunity.
Another molecule expressed on Tregs that contributes to contact-dependent suppression is lymphocyte-activation gene 3 (LAG-3) [21]. In addition to expression on Tregs, LAG-3 is upregulated on the surface of conventional T cells following activation. LAG-3 is associated with the T cell receptor (TCR) on the surface of T cells and binds major histocompatibility complex class II (MHC-II) molecules. This interaction between LAG-3 and MHC-II leads to inhibition of TCR/CD3-mediated T cell activation. LAG-3 can also directly modulate dendritic cell function by interacting with MHC-II and preventing dendritic cell maturation by depriving them of activating signals from conventional CD4+ T cells. Consistent with a role for LAG-3 in contact-dependent suppression by Tregs, in vitro or in vivo blockade of LAG-3 reduces suppression by Tregs [22]. Further, genetic deletion of Lag3 in mice also led to reduced suppressive activity [23]. In melanoma and colorectal cancer patients, LAG-3+ Tregs are expanded in peripheral blood compared with healthy donors and are present at a higher frequency within lymph nodes containing tumor metastases compared with normal lymph nodes [24]. Furthermore, FOXP3+LAG-3+ cells were found to secrete IL-10 and TGF-β and to potently suppress proliferation in a contact-dependent manner in vitro.
A separate contact-dependent mechanism by which Tregs can exert effector function is through the release of cytolytic granules containing granzyme and perforin. Although this is a feature that is normally restricted to CD8+ T cells, Tregs can express granzyme and perforin and can eliminate autologous cells through a perforin-dependent pathway [25]. Tregs expressing granzyme and perforin are therefore able to suppress immune responses through the direct elimination of effector T cells. Finally, one study demonstrated the importance of granzyme and perforin in the suppression of antitumor immunity in a murine cancer model, underscoring the importance of this contact-dependent mechanism in promoting tumor growth [26].
6.3.2 Suppression of Immune Responses via Soluble Factors
The second general mechanism by which Tregs can exert their suppressive function is either by secretion, uptake, or generation of soluble molecules. As discussed earlier, Tregs are characterized by constitutive expression of the IL-2 receptor CD25. This high expression of CD25 causes Tregs to preferentially bind IL-2, depriving conventional T cells of this important stimulatory cytokine. Effector T cell deprivation of IL-2 at inflammatory sites subsequently leads to loss of their effector function and apoptosis [27].
A second soluble mechanism used by Tregs to suppress immune responses is the secretion of cytokines, such as interleukin-10 (IL-10) [28]. In particular, IL-10 production by Tregs plays a role in controlling inflammation at mucosal sites, and mice that lack the Il10 gene in Tregs develop spontaneous colitis and inflammation of the skin and lungs [29]. IL-10 can directly inhibit effector T cells through interaction with the hetero-tetrameric IL-10 receptor complex, leading to activation of STAT3 and transcription of anti-inflammatory genes [30]. In addition to direct suppression of effector T cells, IL-10 can also suppress immune responses by limiting the ability of macrophages to produce inflammatory cytokines [31]. Similarly, IL-10 also prevents the maturation of dendritic cells and inhibits their expression of co-stimulatory molecules [32]. While Tregs are noted for their ability to secrete IL-10, other cell types, such as macrophages under certain conditions, also secrete IL-10 [28]. Although IL-10 is a highly pleiotropic cytokine, it has a clear role in suppression of immune responses by Tregs.
Another important cytokine produced by Tregs that has been shown to play a broad and important role in the immune system is TGF-β [33]. Unlike other cytokines, TGF-β is initially translated as an inactive protein that requires proteolysis for activation. Inactive TGF-β is non-covalently bound to latency-associated peptide (LAP) through an association with GARP on the surface of Tregs [34]. This membrane-bound form of TGF-β is then activated through several possible proteolytic pathways, allowing the activated form of TGF-β to perform its immunosuppressive function [35]. One of the first descriptions of a connection between TGF-β and Tregs was in a model of experimental autoimmune encephalitis (EAE) in mice, where oral tolerance was induced by feeding mice myelin basic protein [36]. Analysis of the CD4+ T cells that infiltrated the nervous system to facilitate tolerance revealed that these cells produced TGF-β and prevented EAE. As with other Treg molecules, knockout of TGF-β from murine Tregs leads to induction of autoimmune disease, underscoring the importance of TGF-β in immune homeostasis [37]. TGF-β suppresses effector T cell responses in several ways, including inhibiting IL-2 production and IFN-γ and perforin production in CD8+ T cells [38]. In head and neck cancer patients, an important role of TGF-β secreting Tregs has been described [39]. Taken together, secretion of TGF-β by Tregs plays an important role in maintaining immune homeostasis and can inhibit antitumor immunity.
Another important cytokine produced by Tregs to facilitate immunosuppression in murine models is interleukin-35 (IL-35) [40]. IL-35 is a member of the IL-12 family of heterodimeric cytokines and consists of one IL-12α subunit and one IL-27β/Ebi3 (Epstein-Barr virus-induced gene 3) subunit [41]. These cytokine genes are constitutively expressed in a subpopulation of murine Tregs, but not conventional T cells, and are upregulated following Treg activation. IL-35 confers suppressive activity on naïve CD4+ T cells and directly suppresses division of conventional cells. Like other inhibitory cytokines, IL-35 can also drive the development of an induced Treg population, called iTr35, that can suppress effector T cells via IL-35 [42]. IL-35 mediates signaling via a unique IL12rβ2:gp130 receptor heterodimer and a STAT1:STAT4 heterodimer [43]. In murine cancer models, IL-35 has recently been shown to play an important role in promoting tumor growth by contributing to T cell exhaustion in the tumor microenvironment [44]. Consistent with increased IL-35 production from highly activated Tregs, an IL-35 reporter mouse revealed enrichment of IL-35+ Tregs in the tumor microenvironment, and neutralization of IL-35 or Treg-specific genetic deletion of Ebi3 led to enhanced antitumor immunity, which was mediated via enhanced cell proliferation and effector function and improved memory cell generation of effector T cells. Treg-restricted deletion of Ebi3 also led to reduced expression of the inhibitory receptors PD-1, LAG-3, and TIM-3, suggesting that IL-35 may promote exhaustion through upregulation of multiple inhibitory receptors [44].
Finally, Tregs can also mediate immunosuppression via the generation of adenosine, a labile, highly suppressive molecule [45]. Extracellular adenosine accumulates at sites of ischemia and inflammation in vivo. In the extracellular space, adenosine is generated by Tregs via breakdown of ATP. The extracellular ectoenzymes CD39 and CD73 on Tregs, or cells in close proximity, in tandem catalyze the breakdown of ATP to adenosine. While CD73 is broadly expressed on activated T cells and other cell types, CD39 expression is largely restricted to Tregs. Increased levels of adenosine at sites of inflammation inhibit immune responses through interaction with either the adenosine A2A receptor on effector T cells or the adenosine A2B receptor on antigen-presenting cells. The interaction of extracellular adenosine with either receptor leads to increased intracellular levels of cAMP and limits the release of inflammatory cytokines from both effector T cells and antigen-presenting cells. Tregs can therefore limit the production of inflammatory cytokines locally by breaking down ATP into adenosine through extracellular ectoenzymes.
As described in this section, Tregs use multiple contact-dependent and contact-independent/contact-soluble mechanisms to suppress effector T cell responses and antigen-presenting cell development and function. Given the detrimental effects of autoimmunity and excessive immune responses and the diversity of cell populations and effector mechanisms they need to control, Tregs likely have evolved multiple immunosuppressive mechanisms to adequately control autoimmunity and inflammation in a variety of settings. An important question is whether certain mechanisms are more dominantly or preferentially utilized by Tregs in tumors and thus may be targeted therapeutically without substantially impacting the ability of Tregs to maintain immune homeostasis and peripheral tolerance.
6.3.3 Potentiation of Suppression and Survival
Tregs function in diverse environments and suppress a variety of cell types. Consequently, their function and survival are likely modulated or potentiated by a variety of environmental cues, many of which are likely poorly understood or have yet to be defined. Early studies suggested that Treg suppression was contact dependent [16, 17]. However, this notion was inconsistent with the growing appreciation of the importance of cytokines in mediating Treg-dependent suppression. This conundrum was resolved when a more recent study showed that it was not suppression by Tregs per se that was exclusively contact dependent but rather the boosting/potentiation of their suppressive activity that was contact dependent [46]. This study found that co-culture of Tregs with fixed or live conventional CD4+ T cells or antigen-presenting cells was sufficient to boost the capacity of Tregs to suppress effector T cells across a permeable transwell membrane via IL-10 and IL-35.
The potentiation of Treg function and survival was found to be mediated by neuropilin-1 (Nrp1) on the surface of Tregs via interaction with Sema4a [47]. Nrp1 is involved in normal neural and vascular development and also plays a role in tumor angiogenesis [48]. Signaling through the Nrp1/Sema4a interaction is necessary for Treg suppression by soluble cytokines in vitro [47]. Nrp1 on Tregs limits Akt (protein kinase B [PKB]) activity via phosphatase and tensin homolog (PTEN), which in turn stabilizes the Treg phenotype and enhances their survival and function. Consistent with a requirement for Nrp1 to mediate Treg potentiation, genetic ablation of Nrp1 in murine Tregs led to a significant enhancement of antitumor immunity in vivo but did not lead to overt autoimmunity or peripheral inflammation [47, 49]. These observations highlighted the unique role of Nrp1 in stabilizing and potentiating the survival and suppressive function of Tregs in the tumor microenvironment. However, it remains to be determined if the Nrp1 pathway is only utilized in the tumor microenvironment and if so why and if there are other mechanisms that regulate Treg fate and function.
6.4 Relationship Between Regulatory T Cells and Cancer
Tregs are indispensable in vivo for their control of immune homeostasis through suppression of autoreactive T cells. Tumor tissue originates from healthy tissue, and as such Treg suppression of autoreactive immune responses likely limits antitumor immune responses because of their normal role in protecting tissue from damage caused by overt inflammation. Experimental evidence from murine models has highlighted the importance of Tregs in suppressing antitumor immunity, while the presence of Tregs in human tumors correlates with poor prognosis.
6.4.1 Role of Regulatory T Cells in Suppression of Antitumor Immunity
The role that Tregs play in the suppression of antitumor immunity has been demonstrated in several mouse models. Mutation or elimination of the Foxp3 gene in mice and humans leads to fatal autoimmunity, so germline deletion of Foxp3 in mice cannot be used to study antitumor immunity. While Tregs can be limited or depleted in adult mice with antibodies targeting CD4 or CD25 or drugs such as cyclophosphamide, these treatments also impact activated effector T cells, confounding experimental interpretation [50]. Instead, inducible Treg-targeting genetic systems have been used to transiently deplete Tregs in adult mice to study their role in suppressing antitumor immunity. One model that has been utilized in mice is insertion of the human diphtheria toxin receptor (DTR) under control of the Foxp3 locus (Foxp3 DTR) [51, 52]. Following the administration of diphtheria toxin, all cells expressing FOXP3 are depleted, allowing for a direct assessment of the role of Tregs in suppressing antitumor immunity.
Using the Foxp3 DTR mice, depletion of FOXP3+ Tregs has been performed in mice with a variety of implanted tumors. Following depletion of Tregs, mice had reduced tumor growth and prolonged survival compared with littermates that did not have their Tregs depleted [47, 53]. These mechanistic studies in mice have demonstrated that Tregs play an important role in suppressing antitumor immunity and that specific depletion of Tregs is sufficient to prevent tumor growth and prolong survival in mice. However, depletion of Tregs following administration of diphtheria toxin in the Foxp3 DTR mice is not specific for Tregs in the tumor microenvironment, and these mice quickly succumb to autoimmune disease despite their antitumor immune responses. This once again highlights the importance of Tregs in maintaining peripheral tolerance throughout life and suggests that systemic depletion of Tregs may not be a viable treatment option for cancer patients.
6.4.2 Local Expansion of Regulatory T Cells in Tumors
Tregs are present in many healthy tissues, and as tumors grow, Tregs can possibly expand locally through increased antigenic stimulation and subsequent proliferation (Fig. 6.4a). In the context of a tumor, tissue-resident Tregs specific for self-peptides presented on MHC receive additional antigenic stimulation as the tumor grows, leading to expansion of local Tregs. In support of this concept, studies have found that Tregs within tumors have a distinct T cell receptor repertoire from conventional CD4+ T cells [54] and that the T cell receptor repertoire of Tregs is largely skewed toward a few clonally expanded populations [55]. Also, Tregs are often the most proliferative immune cell type in tumors [47]. Expansion of Tregs specific for antigens present in normal tissue may partially explain the increased presence of Tregs within tumors, although this mechanism could exist in conjunction with enhanced trafficking of Tregs to tumors.
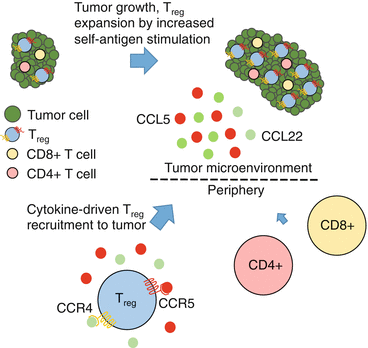
Fig. 6.4
Origins of Tregs in the tumor microenvironment. Tregs can increase in frequency within the tumor microenvironment as the tumor grows by increased self-antigen presentation and subsequent proliferation of Tregs. Enhanced recruitment of Tregs to the tumor microenvironment from the periphery can also occur through interaction of chemokine receptors on Tregs with chemokines produced by tumor cells, tumor-associated macrophages, or CD8+ effector T cells within the tumor microenvironment. This chemokine signaling leads to the preferential accumulation of Tregs within the tumor
6.4.3 Regulatory T Cell Trafficking to Tumor Tissues
To perform their effector function, activated Tregs need to traffic to sites of inflammation within tissues. Trafficking of leukocytes is generally controlled via chemotactic cytokines known as chemokines. These chemokines interact with a specific array of cell surface transmembrane G protein-coupled receptors. For example, under normal physiologic conditions in the lymph node, expression of the chemokine receptor CCR7 on Tregs leads to their recruitment to T cell zones, where they have access to abundant IL-2 [56]. The recruitment of Tregs to T cell zones within lymph nodes highlights the function of chemokine receptors and ligands to target trafficking to specific anatomical locations. Similarly, secretion of specific chemokines by tumors or other immune cells within the tumor microenvironment can actively recruit Tregs through interaction with specific homing receptors on Tregs.
In cancer, specific chemokine/receptor interactions that recruit Tregs to the tumor are dependent on the tissue origin of the tumor and the cytokine milieu produced in the tumor microenvironment. The most commonly reported mechanism by which Tregs are recruited to the tumor microenvironment is through interaction of the chemokine CCL22 with CCR4 expressed on Tregs. First described in breast cancer, this pathway has been found to play an important role in recruiting activated Tregs to other tumor types including ovarian cancer, colorectal cancer, and head and neck cancer [57]. While tumor cells may actively secrete chemokines, tumor-associated macrophages (TAMs) are another major source of chemokines. TAMs were shown to be the major source of chemokines responsible for the recruitment of Tregs in ovarian cancer [58]. CCL22 secretion by tumor-infiltrating CD8+ T cells can also drive Treg recruitment into the tumor [59].
In the normal setting of inflammation, Tregs are recruited to limit tissue damage from the ensuing immune response. This is analogous to recruitment of Tregs to tumors via local expansion or chemokine-mediated recruitment. This highlights the co-opting of a normal biological process to promote tumor-induced tolerance. Studies in a wide variety of murine tumor models have demonstrated that Tregs play a central role in preventing antitumor immunity.
6.4.4 Regulatory T Cells and Prognosis
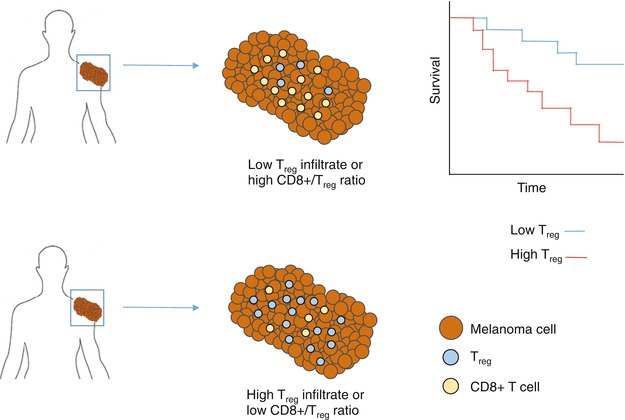
Many studies have evaluated associations between the frequency of Tregs in the tumor microenvironment and clinical outcome, including in head and neck [60], ovarian [58, 62], breast [63], pancreatic [64, 65], gastric [66, 67], lung [68], renal [69, 70], and liver cancers [71] and melanoma [61]. Studies assessing the frequency of Tregs within tumors have generally used tissue sections and have looked for FOXP3+ cells in the presence or absence of additional markers. Other studies have looked at the frequency of Tregs within tumors as a ratio of CD8+ T cells to Tregs [72–75]. A recent meta-analysis of 17 different types of cancer across more than 15,500 cancer cases found that a higher frequency of Tregs in tumors was associated with poorer overall survival when considering all cancer types [76] (Fig. 6.5). Many of these studies used histology of tissue sections and have relied on identifying Tregs using staining for FOXP3 alone or with one or two other markers.
 Peptide-Based Therapeutic Cancer Vaccines
Peptide-Based Therapeutic Cancer Vaccines
 The Human Tumor Microenvironment
The Human Tumor Microenvironment
 Synergy Between Radiotherapy and Immunotherapy
Synergy Between Radiotherapy and Immunotherapy
 Toward Engineered Cells as Transformational and Broadly Available Medicines for the Treatment of Cancer
Toward Engineered Cells as Transformational and Broadly Available Medicines for the Treatment of Cancer
 Plasmacytoid DC/Regulatory T Cell Interactions at the Center of an Immunosuppressive Network in Breast and Ovarian Tumors
Plasmacytoid DC/Regulatory T Cell Interactions at the Center of an Immunosuppressive Network in Breast and Ovarian Tumors
 Immune Monitoring of Blood and Tumor Microenvironment
Immune Monitoring of Blood and Tumor Microenvironment




Related posts:
Peptide-Based Therapeutic Cancer Vaccines
The Human Tumor Microenvironment
Synergy Between Radiotherapy and Immunotherapy
Toward Engineered Cells as Transformational and Broadly Available Medicines for the Treatment of Cancer
Plasmacytoid DC/Regulatory T Cell Interactions at the Center of an Immunosuppressive Network in Breast and Ovarian Tumors
Immune Monitoring of Blood and Tumor Microenvironment
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
