Fig. 2.1
Steroid biosynthetic pathways in the human adrenal cortex. Shown are enzymes (underlined) and intermediates in the biosynthesis of adrenal steroid hormones. 17α-Hydroxypregnenolone is the preferred substrate for the 17,20-lyase reaction of CYP17A1. Consequently, the Δ5 pathway to DHEA is favored over the Δ4 pathway to androstenedione. The adrenal gland produces small quantities of other steroids not shown here. An expanded view of adrenal androgen production is presented later
To initiate steroidogenesis, cholesterol undergoes facilitated transport from a replenishable pool in the outer mitochondrial membrane (OMM) to the inner mitochondrial membrane (IMM), where CYP11A1 (side-chain cleavage enzyme) catalyzes the conversion of cholesterol to pregnenolone [1]. Transcription of the CYP11A1 gene is regulated in a hormonally-responsive manner and determines the net steroidogenic capacity of a cell [3]. Pregnenolone diffuses out of mitochondria and serves as the precursor for the ensuing steps of steroidogenesis, most of which take place in the endoplasmic reticulum (ER) (Fig. 2.2). The final steps of cortisol and aldosterone biosynthesis , catalyzed by the enzymes CYP11B1 and CYP11B2, respectively, occur in mitochondria. Thus, intermediates in the corticoid biosynthetic pathway shuttle between mitochondria and the ER. The electron donors for CYP enzymes in these two cellular compartments are summarized in Table 2.1.
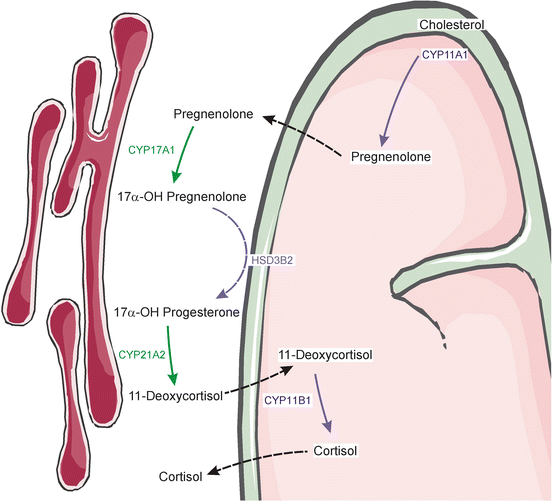
Fig. 2.2
Steroidogenic intermediates shuttle between mitochondria and the ER. The biosynthetic pathway for cortisol is shown; similar shuttling takes place during the synthesis of other adrenal steroid hormones. Enzymatic reactions that occur in mitochondria are shown in purple, whereas those that occur in the ER are in green. Dashed lines indicate passive diffusion across mitochondrial membranes. Prepared using image vectors from Servier Medical Art (www.servier.com), licensed under the Creative Commons Attribution 3.0 Unported License (http://creativecommons.org/license/by/3.0/)
Table 2.1
Cytochrome P450 enzymes involved in adrenal steroidogenesis
CYP classification | Location | Enzyme | Electron donor |
|---|---|---|---|
Type I | Mitochondria | CYP11A1 | NADPH via a flavoprotein (ferredoxin reductase) and an iron-sulfur protein (ferredoxin) |
CYP11B1 | |||
CYP11B2 | |||
Type II | ER | CYP17A1 | NADPH via a flavoprotein [P450-oxidoreductase (POR)] |
CYP21A2 |
Zones of the Adrenal Cortex
In both the fetus and adult, the adrenal cortex is divided into concentric zones that produce different classes of steroid hormones [4, 5].
Human Fetal Adrenal Cortex
At the eighth week of human gestation, the fetal adrenal cortex comprises two morphologically distinct layers: an outer definitive zone (Dz) and an inner fetal zone (Fz) [6]. The Dz is thin and contains small basophilic cells, whereas the Fz is thick and contains large eosinophilic cells (Fig. 2.3). The Dz does not synthesize significant amounts of steroid hormones, but the Fz produces large quantities of DHEA and its sulfated counterpart DHEA-S. Cells of the Fz express CYP17A1, a dual function enzyme that catalyzes both a 17α-hydroxylation reaction and a 17,20-lyase reaction required for C19 steroid production [1]. The lyase reaction is selectively enhanced through allosteric interactions with cytochrome b5 (CYB5), a protein that is abundant in the Fz [1]. A third cortical zone, the transitional zone (Tz), develops shortly after the appearance of the Fz and Dz. The Tz secretes cortisol, a hormone that promotes maturation of the lungs and other organs [8].
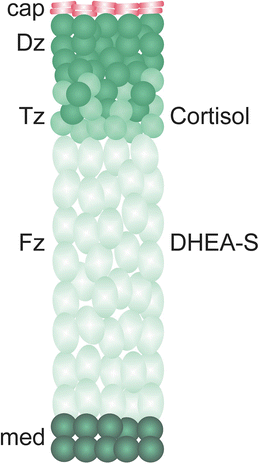
Fig. 2.3
Structure of the human fetal adrenal gland. The zones of the fetal cortex are the Dz, Tz, and Fz. The Tz and Fz produce cortisol and C19 androgen precursors, respectively. An early burst of cortisol production by the Tz during the 9th week of gestation, coinciding with a transient increase in expression of HSD3B2, is thought to safeguard female sexual development by limiting the production of androgen precursors by the Fz [7]. After birth the Dz differentiates into the functionally distinct zones of the adult cortex. Cap capsule, DHEA–S dehydroepiandrosterone sulfate, Dz definitive zone, Fz fetal zone, med medulla, Tz transitional zone
C19 steroids secreted by the Fz are converted into estrogens through the actions of enzymes in the liver and/or placenta. The fetal pituitary, adrenal, liver, and placenta constitute a functional entity known the feto-placental unit [9] (Fig. 2.4). The concentration of estrogens in maternal plasma increases abruptly mid-gestation, reflecting production by this unit [10]. Estrogens support pregnancy by promoting maternal breast development, blood volume expansion, and uterine growth/contractility, although intact fetal adrenocortical function is not a prerequisite for term gestation or birth [11].
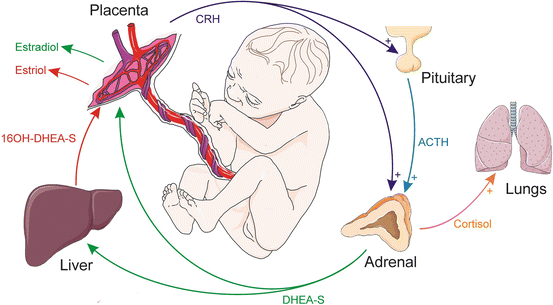
Fig. 2.4
Steroid production by the feto-placental unit . Placental CRH and pituitary-derived ACTH promote cortisol and DHEA-S secretion by the fetal adrenal gland. DHEA-S is converted into estrogens (estradiol and estriol) by enzymes in the liver and placenta. The resultant estrogens support pregnancy, while cortisol promotes the maturation of the lungs and other organs in the fetus. ACTH adrenocorticotropic hormone, CRH corticotropin-releasing hormone, DHEA-S dehydroepiandrosterone sulfate, 16OH-DHEA-S 16-hydroxydehydroepiandrosterone sulfate. Prepared using image vectors from Servier Medical Art (www.servier.com), licensed under the Creative Commons Attribution 3.0 Unported License (http://creativecommons.org/license/by/3.0/)
Adrenocorticotropic hormone (ACTH), a peptide secreted by the anterior pituitary gland, is a major regulator of fetal adrenal growth and function. ACTH promotes the production of both C19 steroids and cortisol in the fetal adrenal. Disruption of hypothalamic/pituitary function (e.g., in the anencephalic fetus) impairs Fz growth and decreases estrogen levels in the maternal circulation [8].
Another important regulator of steroidogenesis in the fetus is placenta-derived corticotropin-releasing hormone (CRH), a peptide that both directly and indirectly stimulates fetal adrenal cells to produce cortisol and androgens [12, 13] (Fig. 2.4). Unlike hypothalamic CRH secretion, which is subject to feedback inhibition by glucocorticoids (see later), placental CRH release is enhanced by cortisol. At birth the placenta separates from the body, and this CRH feed-forward loop is interrupted. The newborn adrenal gland, which is almost as large as the kidney, shrinks dramatically over the first 2 weeks of life owing to apoptotic involution of the Fz . Regression of the Fz is accompanied by a reduction in C19 steroid production [1]. Postnatally, the Dz differentiates into the anatomically and functionally distinct zones of the adult cortex.
Human Adult Adrenal Cortex
The adult adrenal cortex of humans contains three layers: the zona glomerulosa (zG), zona fasciculata (zF), and zona reticularis (zR) (Fig. 2.5) [4, 14]. The cortex is enveloped by a capsule that provides structural support and houses stem/progenitor cells of the cortex. A complex network of blood vessels within the adrenal gland ensures the efficient delivery of stimulants and the export of corticoids into the circulation [15, 16]. Blood flows centripetally in the gland, so inner cortical zones and the adrenal medulla are exposed to high concentrations of locally-produced steroids.
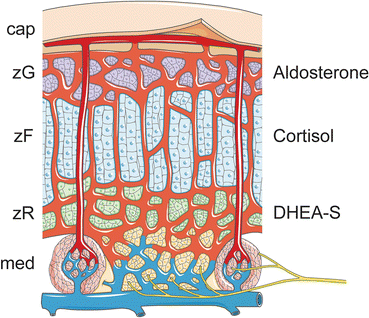
Fig. 2.5
Structure of the adult adrenal gland. The gland is covered by a capsule. The cortical zones are the zG, zF, and zR, which produce aldosterone, cortisol, and DHEA-S, respectively. Adrenal glands are highly vascularized, ensuring the efficient delivery of stimulants and export of steroid hormones into the systemic circulation. A subcapsular arteriolar plexus receives oxygenated blood and distributes it to the underlying tissue via two types of vessels: (1) sinusoids that supply the adrenal cortex and medulla and (2) medullary arteries that directly supply to the medulla. The adrenal medulla, which is innervated by preganglionic sympathetic fibers, functions as part of the sympathetic nervous system, releasing catecholamines into the circulation. Cap capsule, DHEA-S dehydroepiandrosterone sulfate, med medulla, zF zona fasciculata, zG zona glomerulosa, zR zona reticularis. Prepared using image vectors from Servier Medical Art (www.servier.com), licensed under the Creative Commons Attribution 3.0 Unported License (http://creativecommons.org/license/by/3.0/)
zG
The outermost zone, the zG, secretes aldosterone. The major physiological stimuli of aldosterone secretion are angiotensin II (Ang II) and extracellular K+, but ACTH also enhances production of this steroid hormone [17]. Aldosterone binds to the mineralocorticoid receptor (MR) in cells of the distal nephron, leading to retention of Na+ and excretion of K+ and H+ by the kidneys [18]. By modulating Na+ balance, aldosterone impacts extracellular fluid volume and blood pressure. Aldosterone also plays important roles in certain cardiovascular, renal, and inflammatory diseases [19–21]. For example, aldosterone exerts direct actions on cardiomyocytes, contributing to cardiac fibrosis and congestive heart failure [22]. Distinctive molecular markers of the zG include CYP11B2 (the mitochondrial enzyme that catalyzes the final steps of aldosterone biosynthesis) and AT1R (the Ang II receptor) [23]. The zG lacks expression of CYP17A1 and therefore is not able to synthesize cortisol or androgen precursors.
zF
The largest cortical zone, the zF, produces the stress hormone, cortisol, as part of the hypothalamic-pituitary-adrenal (HPA) axis (Fig. 2.6). ACTH secreted by the pituitary is the principal stimulus for cortisol production [24]. Cells in the zF respond to ACTH via its G-protein-coupled receptor, the melanocortin 2 receptor (MC2R), working in conjunction with melanocortin 2 receptor accessory proteins (MRAPs). Adrenal-derived cortisol binds to the glucocorticoid receptor (GR), which is expressed in a wide array of cell types including hepatocytes, muscle cells, lymphocytes, neurons, and neuroendocrine cells. Cortisol functions to (1) increase blood glucose concentrations through gluconeogenesis; (2) facilitate the catabolism of proteins, lipids, and carbohydrates; (3) suppress the immune system; and (4) induce enzymes for catecholamine biosynthesis in the medulla [25, 26]. A distinctive molecular marker of the zF is CYP11B1, a mitochondrial enzyme required for the biosynthesis of cortisol [23].
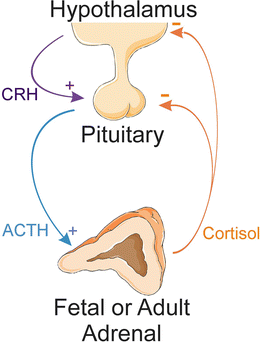
Fig. 2.6
HPA axis . In response to stress or other physiological cues, hypothalamic neurons secrete CRH, which stimulates the release of ACTH from the pituitary gland. ACTH promotes the secretion of cortisol by the adrenal cortex. Cortisol, in turn, inhibits the axis at the level of the pituitary and hypothalamus. ACTH adrenocorticotropic hormone, CRH corticotropin-releasing hormone, HPA hypothalamic-pituitary-adrenal. Prepared using image vectors from Servier Medical Art (www.servier.com), licensed under the Creative Commons Attribution 3.0 Unported License (http://creativecommons.org/license/by/3.0/)
zR
The innermost layer of the cortex, the zR, secr etes the C19 steroids DHEA, DHEA-S, androstenedione, and 11β-hydroxyandrostenedione, which are often termed “adrenal androgens” [1]. DHEA-S is the most abundant adrenal steroid in the circulation of adults [27]. In contrast to more potent androgens, such as testosterone and dihydrotestosterone (DHT), adrenal androgens exhibit little or no binding/activation of the androgen receptor (AR) [28]. Consequently, adrenal C19 steroids function mainly as precursor molecules that can be converted into more potent androgens in peripheral tissues (e.g., hair follicles, sebaceous glands, prostate, and genital skin).
Cells of the zR, like those of the zG and zF, respond to ACTH. In contrast to other zones of the cortex, the zR does not become functionally active until 6–8 years of age (adrenarche), coinciding with increased expression of CYB5, the allosteric regulator that enhances the 17,20-lyase activity of CYP17A1 [1]. CYB5 serves as a distinctive molecular marker of the zR; another characteristic marker of this zone is SULT2A1, the enzyme responsible for sulfation of DHEA [29].
Comparative Adrenal Anatomy of Small Animal Models
Rodents are used as experimental models for the study of adrenal physiology, but there are noteworthy differences between the adrenocortical zonation patterns of rodents and humans [30]. The zR is absent from the adrenal gland of the mouse. The adrenal cortex of the young mouse contains an ephemeral juxtamedullary layer, the X-zone, that regresses during puberty in males and the first pregnancy in females [31]. Lineage tracing studies have shown that the X-zone is a derivative of the fetal cortex [31], but the physiological function of the X-zone remains unclear [32]. The rat adrenal cortex has an additional layer, the undifferentiated zone (zU), located between the zG and zF. The zU contains a transitional population of cells [33].
Species also differ in the complement of steroidogenic enzymes and cofactors expressed in the adrenal cortex, and these differences have biological ramifications. For example, cells in the zF and zR of humans express CYP17A1, so cortisol is the principal glucocorticoid secreted by the adrenal gland of this species [34]. Cells in the zF of adult mice and rats lack CYP17A1, so corticosterone is the main glucocorticoid produced, and adrenal androgens are not made. A newly recognized rodent-like model is the spiny mouse (genus Acomys) [35]. In contrast to the laboratory mouse (genus Mus), the adrenal cortex of the spiny mouse has a zR. Cells in the zF and zR of the spiny mouse produce cortisol and DHEA, respectively. Thus, the adrenal physiology of the spiny mouse recapitulates that of humans [9].
Role of Zonal Remodeling in the Regulation of Steroidogenesis
The adrenal cortex is a dynamic tissue that undergoes continual renewal. Senescent cells are replenished through division and differentiation of stem/progenitor cells in the periphery of the gland (i.e., the adrenal capsule and subcapsule) [36–38]. The newly formed cells migrate centripetally to repopulate cortical zones. This cellular turnover facilitates adrenocortical remodeling in response to physiological demand for steroids. Cortical zones can reversibly expand, contract, or alter their biochemical profiles in response to physiological or pharmacological triggers (Table 2.2). Zonal remodeling is one of the major mechanisms by which steroid production is modulated.
Table 2.2
Impact of various stimuli on adrenocortical remodeling and function
Zone (species) | Physiological or pharmacological stimulus | Effect | References |
|---|---|---|---|
zG (rat) | ↓ Na+ or ↑ K+ in diet | Expands the zone, increasing aldosterone production | |
↑ Na+ or ↓ K+ in diet | Contracts the zone, decreasing aldosterone production | ||
Captopril or other ACE inhibitors | Contracts the zone, decreasing aldosterone production | ||
Endothelin or serotonin | Stimulates aldosterone production | ||
Dopamine or atrial natriuretic peptide | Inhibits aldosterone production | ||
Arginine vasopressin | Expands the zone | ||
zF (rat) | ACTH | Expands the zone, increasing glucocorticoid production | [23] |
Dexamethasone | Contracts the zone, decreasing glucocorticoid production | ||
zR (primates) | Adrenarche | Increase s CYB5 expression, enhancing DHEA production | |
Social status in marmosets | Adult females develop a functional zR based on status in the group | ||
Cortisol and androstenedione | Stimulates DHEA production through competitive inhibition of HSD3B2 activity |
Secreted Peptides Implicated in Adrenocortical Growth and Steroidogenesis
The archetypal peptide hormones impacting adrenocortical function are ACTH and Ang II, but many other hormones and growth factors, working alone or in combination, have been shown to regulate adrenocortical cell function [36, 38] (Table 2.3). Hormones and paracrine factors traditionally associated with reproductive function , such as luteinizing hormone, activins, and inhibins, can affect adrenocortical steroidogenesis.
Table 2.3
Secreted proteins that regulate adrenocortical cell growth and function
Factor | Function | References | |
|---|---|---|---|
Endocrine hormones | ACTH | Stimulates cortisol and androgen biosynthesis; some of its tropic actions are mediated indirectly via growth factors | [8] |
Ang II | Stimulates aldosterone production | [23] | |
Placental CRH | Directly stimulates fetal adrenal cells to produce cortisol and androgens | [13] | |
Human chorionic gonadotropin (hCG) | Drives the growth of the fetal adrenal gland during the first trimester of pregnancy | [8] | |
Luteinizing hormone (LH) | The LH receptor has been shown to be functionally active in the adrenal of adults during pregnancy and other high gonadotropin states | ||
Activins | Inhibit adrenocortical cell growth/survival and modulate steroidogenesis | ||
Inhibins | Inhibit activin signaling in adrenocortical cells | ||
Growth factors | Insulin-related growth factors (IGF1/2) | Promote adrenocortical cell mitosis and survival; enhance the effect of ACTH on steroidogenesis in fetal and adult adrenocortical cells | |
Epidermal growth factor (EGF) | A potent mitogen for cultured fetal and definitive zone cells from mid-gestation human fetal adrenal glands | [49] | |
Fibroblast growth factor-2 (FGF2) | Acts as an adrenocortical cell mitogen; binds to zG cells | [50] | |
Developmental signaling molecules | Sonic hedgehog (SHH) | Ligand secreted by subcapsular cells; promotes steroidogenic differentiation of stem/progenitor cells in the capsule | [51] |
Delta-like homologue-1 (DLK1) | Transmembrane protein that is cleaved and secreted by the rat zU; regulates the differentiation of steroidogenic cell progenitors in the capsule | [33] | |
Wingless-related integration site-4 (WNT4) | Wnt/β-catenin signaling is critical for zG differentiation and maintenance; downregulation of this signaling is required for zG-to-zF conversion | ||
R-spondin-3 (RSPO3) | Ligand that potentiates Wnt/β-catenin signaling and is required for zG differentiation | [54] |
Uptake and Intracellular Trafficking of Cholesterol: Prelude to Steroidogenesis
Initiation of steroidogenesis e ntails the following steps: (1) mobilization of cholesterol, the precursor of all steroid hormones, from endogenous or exogenous sources, (2) transport of cholesterol to the OMM, and (3) transfer of cholesterol from the OMM to the IMM.
Mobilization of Free Cholesterol from Intracellular and Extracellular Sources
Cholesterol can be derived from a combination of sources: (1) de novo biosynthesis, (2) import of lipoprotein-associated cholesteryl esters (CEs) via endocytosis of the low-density lipoprotein receptor (LDLR), (3) uptake of esterified cholesterol through the high-density lipoprotein (HDL) scavenger receptor 1 (SR-B1), and (4) hydrolysis of CEs stored with lipid droplets. These redundant sources ensure that adequate cholesterol is available for steroidogenesis.
De Novo Cholesterol Biosynthesis
Cholesterol is synthesized from mevalonate and isoprenoid precursors [55]. The rate-limiting step of cholesterol biosynthesis is catalyzed by hydroxymethylglutaryl coenzyme A (HMG-CoA) reductase. Expression of HMG-CoA reductase is regulated by isoforms of sterol-regulatory element-binding protein (SREBP), a transcription factor that coordinate the synthesis, uptake, and metabolism of cholesterol and fatty acids [56]. Newly synthesized SREBP is membrane-bound and resides in the ER, where it interacts with SREBP cleavage-activating protein (SCAP), a key cellular cholesterol sensor [57] (Fig. 2.7). When cells are deficient in cholesterol, SCAP and other proteins escort SREBP from the ER to Golgi. Once in the Golgi, SREBP is proteolytically processed to generate a “mature” transcription factor that travels to the nucleus and activates genes required for de novo cholesterol synthesis (e.g., HMG-CoA reductase) and for internalization of LDL [58]. When cells are replete with cholesterol, SREBP is retained in the ER, thereby limiting its proteolytic activation.
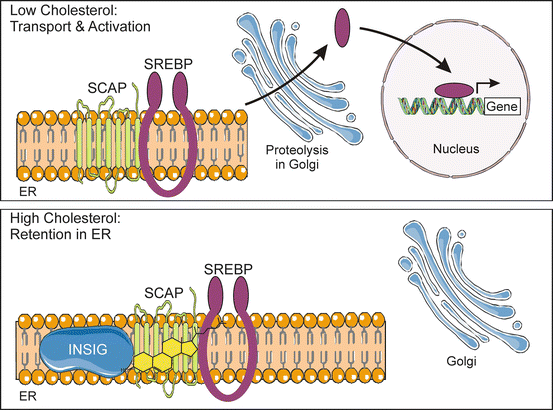
Fig. 2.7
Activation of SREBP , a key transcriptional regulator of steroidogenesis, is regulated by the cholesterol content of the ER membrane. Cholesterol homeostasis is maintained by SREBP, a membrane-associated transcription factor, working in conjunction with SCAP, a cholesterol sensor. When cholesterol levels are low, SCAP and other proteins escort SREBP from the ER to Golgi. Once in the Golgi, SREBP is proteolytically processed to generate a “mature” transcription factor that travels to the nucleus and increases the expression of genes required for cholesterol synthesis and uptake. High levels of cholesterol trigger a conformational change in SCAP, causing the SCAP-SREBP complex to associate with INSIG, an ER anchor protein. Consequently, SREBP is retained in the ER and not proteolytically activated. ER endoplasmic reticulum, INSIG insulin-induced gene, SCAP SREBP cleavage-activating protein, SREBP sterol-regulatory e lement-binding protein. Prepared using image vectors from Servier Medical Art (www.servier.com), licensed under the Creative Commons Attribution 3.0 Unported License (http://creativecommons.org/license/by/3.0/)
De novo synthesis is not the principal source of cholesterol for steroidogenesis, as evidenced by the fact that patients treated with statins, inhibitors of HMG-CoA reductase, exhibit normal cortisol secretion [59, 60]. Nevertheless, de novo synthesis remains an important source of cholesterol under certain physiological and pathophysiological states [61].
Import of Esterified Cholesterol via the LDL Receptor
Approximately 80% of the cholesterol used in the synthesis of adrenal steroids derives from uptake of plasma lipoproteins by LDLR (Fig. 2.8) [62]. Apolipoprotein E (apoE) and apolipoprotein B (apoB)-containing lipoprotein particles bind to LDLR and are internalized via clathrin-coated pits [63]. In late endosomes, lipoprotein-derived CEs are hydrolyzed by lysosomal acid lipase (LIPA) to produce free cholesterol. Loss-of-function mutations in LIPA cause Wolman disease and its milder variant, cholesteryl ester storage disease [64]. The liberation of LDL-derived cholesterol from late endosomes requires the protein products of two other genes NPC1 and NPC2 [65]. NPC2 binds and facilitates the hydrolysis of CEs by LIPA. The resultant-free cholesterol is transferred to NPC1, inserted into the endosomal membrane, and then exported. Mutations in NPC1 or NPC2 cause Niemann-Pick type C disease, another lysosomal storage disorder [66].
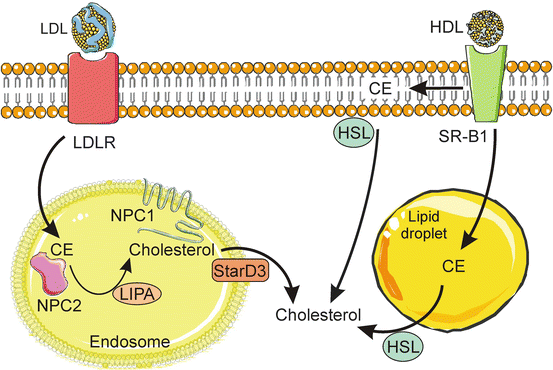
Fig. 2.8
Import of esterified cholesterol via lipoprotein receptors . Adrenocortical cells take up circulating LDL via receptor mediated endocytosis. In late endosomes, LDL-derived CE is bound by NPC2 and hydrolyzed by LIPA to yield free cholesterol, which is then transferred to NPC1, inserted into the endosomal membrane, and exported. A cholesterol-binding protein, StarD3, co-localizes with the NPC system and may participate in the egress of cholesterol from endosomes. SR-B1 mediates the uptake of CE from HDL. Some of the CE delivered via SR-B1 is incorporated into the plasma membrane, and some is directly incorporated into lipid droplets. HDL-derived CE is hydrolyzed to free cholesterol by HSL. CE cholesteryl ester, HDL high-density lipoprotein, HSL hormone-sensitive lipase, LDL low-density lipoprotein, LDLR LDL receptor, LIPA lysosomal acid lipase, NPC1/NPC2 Niemann-Pick type C disease proteins 1 and 2, StarD3 START domain protein 3, SR-B1 scavenger receptor class B, type 1. Prepared using image vectors from Servier Medical Art (www.servier.com), licensed under the Creative Commons Attribution 3.0 Unported License (http://creativecommons.org/license/by/3.0/)
A cholesterol-binding protein, StarD3 (MLN64), co-localizes with NPC proteins and may participate in the egress of LDL-derived cholesterol from endocytic vesicles. StarD3 is a member of the START domain (StarD) family of proteins [67]. The START [StAR (STeroidogenic Acute Regulatory protein)-related lipid Transfer] domain is a conserved sequence that binds sterols and other lipids [68]. The human START domain family comprises 16 proteins, 5 of which (StarD1/D3/D4/D5/D6) bind cholesterol. StarD3 contains a leader sequence that targets the molecule to late endosomes. Enforced expression of a mutant form of StarD3 lacking the START domain causes accumulation of free cholesterol in lysosomes [69]. The roles of other START domain proteins, including the founding member of the family, StarD1 (better known as StAR), are discussed later.
Uptake of Esterified Cholesterol via SR-B1
Scavenger rece ptor class B, type 1 (SR-B1) mediates the uptake of HDL CEs through a mechanism that differs from the endocytic pathway used by the LDL receptor (Fig. 2.8) [70]. SR-B1 localizes to lipid rafts, membrane domains rich in cholesterol and sphingolipids. SR-B1 forms a nonaqueous channel [71]. Binding of HDL to SR-B1 results in the “selective uptake” of esterified cholesterol into the plasma membrane, followed by the release of the resultant lipid-depleted HDL particles back into the circulation. Some CEs delivered via SR-B1 appear to be incorporated directly into lipid droplets [72, 73]. CEs imported into the plasma membrane or lipid droplets by SR-B1 may be hydrolyzed to free cholesterol by hormone-sensitive lipase (HSL). Uptake through SR-B1 is the major source of cholesterol for steroidogenesis in rodents [74]. SR-B1 impacts steroidogenesis in human cell cells, too. Incubation of human HAC15 adrenocortical cells with cholesterol-free synthetic HDL leads to an efflux of cholesterol and a decrease in cortisol production [75].
Hydrolysis of Cholesteryl Esters Stored in Lipid Droplets
Hydrolysis of CEs stored within lipid droplets is the preferred source for cholesterol in the setting of acute hormonal stimulation. HSL, an enzyme activated in response to binding of ACTH or Ang II to their cognate receptors, catalyzes the hydrolysis of lipid droplet-associated CEs [73, 76]. ACTH activates HSL via PKA-mediated phosphorylation, whereas Ang II activates HSL via mitogen-activated protein kinases (MAPKs) (see later). In response to phosphorylation by these kinases, HSL is translocated from the cytoplasm to either the plasma membrane or the surface of lipid droplets, facilitating hydrolysis of CEs at these sites. In addition to catalyzing the hydrolysis of CEs, HSL interacts with various cholesterol-binding proteins to help direct the cholesterol to the OMM for steroidoge nesis [77].
Esterification of Excess Free Cholesterol by SOAT1
Excess-free choleste rol, which is toxic to cells, may be esterified by the ER enzyme sterol O-acyltransferase 1 (SOAT1) [also called acyl-CoA:cholesterol O-acyltransferase 1 (ACAT1)]. The resultant CEs may be incorporated into lipid droplets for storage. Drugs that inhibit SOAT1 activity, such as mitotane and ATR-101, induce the accumulation of cholesterol and free fatty acids in the ER of adrenocortical cells (Fig. 2.9) [78–80]. This increase in cholesterol limits SREBP activation, resulting in downregulation of target genes that mediate cholesterol synthesis and uptake. The accumulation of cholesterol and free fatty acids induces ER stress, which triggers transcription of unfolded protein response (UPR) genes [81]. Persistent ER stress leads to upregulation of pro-apoptotic BAX and repression anti-apoptotic BCL2, thus inducing cell death [79]. Targeted deletion of SOAT1 in an adrenocortical cell line recapitulates the effects of pharmacological inhibitors of SOAT1 [78]. Inhibition of cholesterol esterification is thought to be the mechanism of action of mitotane, the only drug approved for the treatment of patients with adrenocortical carcinoma (ACC) (Fig. 2.9). ATR-101 is undergoing preclinical and clinical testing as a new drug for the treatment of ACC [78].
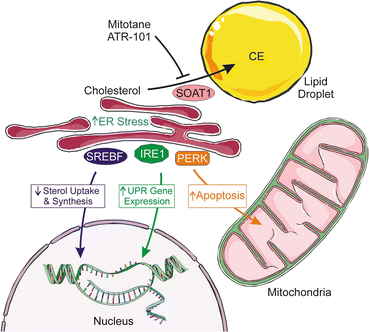
Fig. 2.9
Pharmacological inhibition of SOAT1, the enzyme that esterifies excess cholesterol, impairs adrenocortical steroidogenesis and cell survival. Inhibition of SOAT1 by either mitotane or ATR-101 leads to an increase in free cholesterol and fatty acids that (1) cause downregulation of SREBF-dependent genes that mediate cholesterol synthesis and uptake and (2) trigger ER stress, leading to IRE1-dependent splicing of XBP1 mRNA and subsequent transcription of UPR genes. As ER stress persists, increased expression of PERK induces increased CHOP expression, which triggers expression of pro-apoptotic BAX and repression of anti-apoptotic BCL2, thus inducing cell apoptosis. CE cholesteryl ester, CHOP CCAAT-enhancer-binding protein homologous protein, ER endoplasmic reticulum, PERK PKR-like ER kinase, SREBF sterol-regulatory element-binding protein, SOAT1 sterol O-acyltransferase 1, UPR unfolded protein response, XBP1 X-box-binding protein. Prepared using image vectors from Servier Medical Art (www.servier.com), licensed under the Creative Commons Attribution 3.0 Unported License (http://creativecommons.org/license/by/3.0/)
Transport of Free Cholesterol to the OMM
Mitochondria contain a limited amount of cholesterol, the majority of which resides in the OMM [82, 83]. Therefore, cholesterol consumed during steroidogenesis must be continually replenished. Mitochondrial stores cannot be restocked with cholesterol via simple diffusion across aqueous cytoplasmic spaces, because this compound is relatively insoluble in water. Instead, “free” cholesterol is trafficked to mitochondria via two general mechanisms: (1) vesicular transport, an energy-dependent process entailing budding and fusion of membrane vesicles, and (2) non-vesicular transport via soluble cholesterol-binding proteins [61, 71]. Close physical interactions between mitochondria and other subcellular compartments (e.g., ER, lipid droplets) promote cholesterol transfer.
Role of the Cytoskeleton in Cholesterol Delivery to the OMM
Intracellular vesicular trafficking of cholesterol requires rearrangement of an integrated network of cytoskeletal elements, composed of intermediate filaments, microtubules, and other structures [84–86]. Some cytoskeletal rearrangements appear to be driven by the protein myristoylated alanine-rich C-kinase substrate (MARCKS), which is phosphorylated in adrenocortical cells in response to hormonal stimulation [87] and can associate with actin filaments and the membrane surfaces [88]. Another key cytoskeletal protein is the intermediate filament vimentin, which binds to lipid droplets and to HSL and appears to regulate cholesterol delivery to mitochondria. Pharmacological or genetic disruption of cytoskeletal structure impacts cholesterol trafficking. For example, vimentin null mice exhibit decreased ACTH-stimulated corticosterone levels [89].
Role of SNARE Proteins in Cholesterol Delivery to Mitochondria
SNARE proteins [an acronym derived from “SNAP (Soluble N-ethylmaleimide-sensitive factor Attachment Protein) REceptor”] are GTP-binding proteins that mediate vesicle fusion [90]. The most extensively studied SNARE proteins are those that mediate the fusion of synaptic vesicles with the presynaptic membrane in neurons, but recent studies suggest that these proteins also regulate cholesterol transfer [91]. Certain SNARE proteins, including α-SNAP and SNAP25, are associated with lipid droplets [92]. Gene-silencing experiments and in vitro reconstitution assays have shown that specific SNARE proteins [α-SNAP, SNAP25, syntaxin (STX)-5, and STX-17] are required for efficient cholesterol movement to mitochondria for steroidogenesis [91]. A subset of SNAREs, including STX-5, exhibit cholesterol-binding properties, and this may aid in the trafficking of cholesterol from lipid droplets to the OMM [93, 94].
MAM-Facilitated Transfer of Cholesterol to the OMM
Trafficking of cholesterol to mitochondria is also facilitated by the mitochondria-associated ER membrane (MAM), a distinct subdomain of the ER that is reversibly tethered to the OMM by lipids and protein filaments [71, 95, 96]. MAMs serve as hubs that coordinate metabolic interactions between these two organelles. MAMs have been implicated in a wide array of cellular processes, including lipid transport (e.g., mitochondrial import of phosphatidylserine), Ca2+ signaling, energy metabolism, mitochondrial dynamics, and apoptosis [97, 98].
MAMs are composed of cholesterol-rich lipid rafts and resident membrane proteins (Fig. 2.10). MAM proteins include: (1) mitofusin 2 (MTF2), which functions as a tether; (2) FATE1, which modulates ER-mitochondrial distance [99]; (3) the voltage-dependent anion channel (VDAC), an OMM-associated protein that allows for the passage of ions, metabolites, and signaling molecules (e.g., ATP) across the membrane (see later); and (4) sigma-1 receptor, which promotes cholesterol compartmentalization and trafficking [100, 101]. It has been proposed that cholesterol transfer from OMM to IMM occurs at specialized contact sites bridging the two membranes composed of VDAC and IMM adenine nucleotide translocase (ANT).
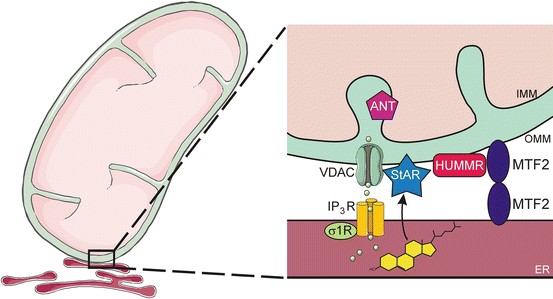
Fig. 2.10
Mitochondria-associated ER membrane (MAM). The ER communicates with mitochondria via the MAM, a specialized subdomain of the ER with the characteristics of a lipid raft. MAMs serve to coordinate metabolic interactions (e.g., lipid transport, Ca2+ signaling) between these two organelles. The following MAM components are illustrated: MTF2 (a tether), VDAC (a channel), σ-1 (a receptor), StAR (a transport protein), HUMMR (an OMM adaptor protein), ANT (an IMM nucleotide translocase), and IP3R, (a receptor and Ca2+ channel). ANT adenine nucleotide translocase, ER endoplasmic reticulum, IMM inner mitochondrial membrane, HUMMR hypoxia-upregulated mitochondrial movement regulator, IP 3 R inositol (1,4,5) trisphosphate receptor MTF2, mitofusin 2, OMM outer mitochondrial membrane, σ1R sigma-1 receptor, and VDAC voltage-dependent anion channel. Prepared using image vectors from Servier Medical Art (www.servier.com), licensed under the Creative Commons Attribution 3.0 Unported License (http://creativecommons.org/license/by/3.0/)
Genetic evidence supporting a role for the MAM in steroidogenesis has emerged from the isolation and characterization of mutant cell lines with altered intracellular cholesterol homeostasis. One such cell line harbors a mutation in the H/ACA small nucleolar RNA (snoRNA) U17, which regulates hypoxia-upregulated mitochondrial movement regulator (HUMMR), an OMM adaptor protein that promotes the formation of ER/mitochondrial contact sites [102].
Role of StarD4/D5/D6 in Cholesterol Trafficking
As mentioned earlier, there are five START proteins in the human genome that can bind cholesterol. Two of these have leader peptides that direct them to specific organelles: StarD1 (StAR itself), which is targeted to mitochondria, and StarD3, which is targeted to endosomes (see later). The remaining members of this subgroup—StarD4, StarD5, and StarD6—lack leader peptides and appear to be cytosolic proteins involved in non-vesicular cholesterol transport in various cell types, including steroidogenic cells. StarD4 is thought to be the principal agent delivering cholesterol to the OMM from elsewhere in the cell [67]. StarD5 binds cholesterol and bile acids, whereas StarD6 binds cholesterol and steroid hormones.
Transfer of Cholesterol from the OMM to the IMM
Once cholesterol reaches the OMM, a group of proteins led by steroidogenic acute regulatory protein (StAR) facilitate the transport of cholesterol to the IMM, the site of pregnenolone synthesis by CYP11A1. The delivery of cholesterol from the OMM to the IMM is the rate-limiting step in steroid production in adrenocortical cells. Hormones, such as ACTH and Ang II, rapidly stimulate this process. Transport occurs preferentially at sites of close contact between the OMM and IMM, and such sites are more plentiful in tubulovesicular than lamelliform mitochondria. The preponderance of tubulovesicular mitochondria in the zF compared to the zG reflects higher levels of steroidogenesis in the former [23].
StAR
The acute response to hormone stimulation is characterized by a rapid increase in the rate of steroid hormone biosynthesis, and classic studies indicated that the induction of steroidogenesis requires new protein synthesis (i.e., is cycloheximide-sensitive) [103, 104]. The acute steroidogenic response is controlled by StAR (StarD1), a rapidly synthesized and short-lived phosphoprotein [105, 106]. The principal function of StAR is to move cholesterol from the OMM to the IMM.
The expression and activation of StAR are tightly regulated by various signaling kinases, including PKA, protein kinase C (PKC), and extracellular signal-regulated kinases 1 and 2 (ERK1/2) [107, 108]. In response to hormonal stimulation, StAR is phosphorylated [109, 110], which enhances its activity [111]. Hormonal stimulation also modulates the activity of various transcription factors that regulate StAR expression [112, 113] (Fig. 2.11). For example, PKA-mediated activation of HSL leads to increased production of not only free cholesterol but also oxysterols, which activate liver X receptors (LXRs) that upregulate StAR transcription in a feed-forward manner for steroidogenesis [114, 115].
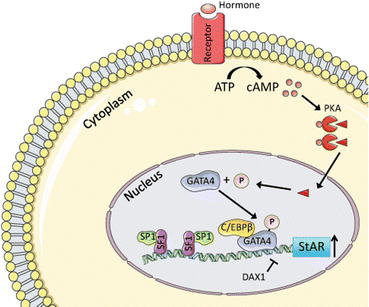
Fig. 2.11
Transcriptional regulation of StAR expression in steroidogenic cells. Shown is the regulation of StAR expression in testicular Leydig cells; similar mechanisms operate in fetal and adult adrenocortical cells. Hormonal stimulation leads to increases in the level of cAMP. Binding of cAMP to the regulatory subunit of PKA (PRKAR1A) allows dissociation of the catalytic subunit (triangle) and its translocation to the nucleus, where it phosphorylates target transcription factors, including GATA4. SF1 is essential for basal StAR expression and binds another widely expressed transcription factor, Sp1. CCAAT-enhancer-binding protein (C/EBPβ) acts synergistically with GATA4. DAX1 functions as a repressor. Prepared using image vectors from Servier Medical Art (www.servier.com), licensed under the Creative Commons Attribution 3.0 Unported License (http://creativecommons.org/license/by/3.0/)
The StAR gene encodes a 37 kDa “precursor” protein that harbors a mitochondrial targeting sequence [108, 116]. When StAR engages the OMM, portions of the molecule become membrane-associated, and StAR undergoes a conformational change that allows it to bind and release cholesterol [117]. As StAR is imported into the mitochondria, it undergoes proteolytic processing from a 37 kDa to a “mature” 30 kDa form [3]. This 30 kDa form localizes to the mitochondrial matrix and retains full StAR activity. Each molecule of StAR is thought to recirculate across the mitochondrial membranes many times, delivering up to 400 molecules of cholesterol to the IMM [118]. To prevent mitochondrial impairment from excess StAR accumulation, the 30 kDa form of the molecule is eventually degraded by proteases in the mitochondrial matrix [119]. Acute mitochondrial accumulation of StAR provokes a retrograde mitochondrial-nuclear signaling and transcriptional upregulation of the proteases involved in the degradation of StAR [119].
The importance of StAR for steroidogenesis is underscored by the disease lipoid congenital adrenal hyperplasia (lipoid CAH), an autosomal recessive disorder caused by loss-of-function mutations in StAR [120, 121]. Lipoid CAH is characterized by enlarged, lipid-laden adrenal glands that make minimal steroids. The condition is lethal in the neonatal period unless promptly diagnosed and treated with corticosteroids. Targeted ablation of the Star gene in mice recapitulates the phenotype of lipoid CAH in humans [122–124].
StAR’s Entourage
StAR interacts with a variety of membrane-associated proteins. It has been proposed that these proteins are organized into a dynamic complex termed the transduceosome, which functions to translocate cholesterol efficiently to the IMM [125]. In addition to StAR, the transduceosome contains VDAC, the translocator protein (TSPO), a TSPO-associated protein PAP7, and other proteins [126]. In steroidogenic tissues that do not express StAR, such as the placenta, the remaining components of the transduceosome, working in conjunction with StarD3, may provide basal cholesterol transport to the IMM for production of pregnenolone.
Voltage-Dependent Anion Channel
VDAC is an OMM channel-forming protein that regulates the passage of ions and small molecules through the OMM. This function determines membrane potential, thereby regulating cell metabolism [127]. There are three isoforms of VDAC, termed VDAC-1, VDAC-2, and VDAC-3 [95]. In addition to controlling metabolism, VDAC isoforms influence programmed cell death by facilitating the release of apoptotic proteins located in the intermembranous space and serving as the proposed target of pro- and anti-apoptotic members of the BCL2 family [127].
VDAC, acting in conjunction with the σ-1 receptor in the MAM, helps facilitate cholesterol transfer for the initiation of pregnenolone synthesis [101]. VDAC interacts with the 37 kDa StAR precursor at the OMM and controls proteolytic processing, allowing the translocation of StAR into mitochondria as a mature 30 kDa protein [96, 128].
Translocator Protein: Necessity is the Mother of Contention
Translocator protein (TSPO), also known as the peripheral benzodiazepine receptor (PBR), is a highly conserved OMM protein [129]. TSPO is expressed in most mammalian tissues and found at highest levels in steroid-producing cells. Early evidence suggested that TSPO supports steroidogenesis by modulating cholesterol transport into mitochondria. The TSPO/PBR ligand PK11195 was shown to stimulate steroidogenesis in both adrenocortical and Leydig cell lines [130]. Mutagenesis and modeling studies indicated that TSPO could bind cholesterol with high affinity [131, 132]. TSPO and StAR were shown to be closely associated in fluorescence energy transfer experiments [133].
Recent experiments, however, have challenged the necessity of TSPO for steroidogenesis, inciting contentious debate [95, 129, 134–136]. Gene targeting studies in cell lines and mice have yielded conflicting results, ranging from no effect on steroidogenesis to impairment of ACTH-stimulated glucocorticoid production [135, 137–140]. The precise function of TSPO remains an area of active investigation [141].
PAP7/ACBD3
PBR-associated protein 7 (PAP7) was identified in a yeast-two hybrid screen employing TSPO/PBR as bait [142]. In steroidogenic cells, PAP7 binds TSPO and the PKA regulatory subunit 1α (PKAR1A) [142, 143]. The PKA holoenzyme comprises two regulatory subunits and two catalytic subunits. Binding of cAMP to the regulatory subunits of PKA triggers the release of catalytic subunits, thereby activating the enzyme. PAP7 has been proposed to function as a kinase-anchoring protein that recruits and confines the PKA holoenzyme at key intracellular sites. A rise in cAMP leads to local release of the catalytic subunits, which phosphorylate StAR present at the OMM [144]. In addition to binding PKAR1A, PAP7 contains an acyl-coenzyme A (CoA)-binding domain, prompting its renaming to acyl-CoA-binding domain containing protein 3 (ACBD3).
Regulation of Cortisol Secretion
Glucocorticoids are essential for basal homeostasis and the response to stress, so production of cortisol is tightly regulated by HPA axis and locally acting factors.
ACTH Synthesis and Release
ACTH , a 39-amino acid peptide secreted by the anterior pituitary, is the principal stimulus for cortisol production [24]. When the HPA axis is activated by stress or other stimuli, neurons in the hypothalamic paraventricular nucleus secrete CRH and arginine vasopressin (AVP) into the hypophyseal portal circulation [145, 146]. CRH is the main stimulant of ACTH release. AVP acts synergistically with CRH to enhance ACTH secretion, although AVP is ineffective in the absence of CRH [147].
ACTH is derived from pro-opiomelanocortin (POMC), a 214-amino acid precursor synthesized in the anterior pituitary and limited other sites [148]. POMC can be proteolytically processed to generate ACTH and several other peptides, including α-melanocyte-stimulating hormone (α-MSH) and β-endorphin [149]. The N-terminal 18 amino acids of ACTH confer its biological activity, so shorter synthetic peptides [ACTH (1–24) and ACTH (1–18)] are used clinically in lieu of full-length ACTH. The sequence of α-MSH, which is contained within the ACTH peptide, stimulates the production of melanin by melanocytes, resulting in skin hyperpigmentation when secreted in excess, as in the ACTH-dependent Cushing syndrome.
ACTH promotes steroidogenesis at three levels: (1) acute ACTH exposure rapidly (within minutes) stimulates the transport of cholesterol from the OMM to the IMM, (2) longer term exposure to ACTH (hours) promotes transcription of genes encoding steroidogenic enzymes, notably CYP11A1, and (3) prolonged exposure to ACTH (weeks to months) promotes zonal expansion and adrenal growth [126, 150].
Feedback Inhibition of the HPA Axis
Cortisol is the main negative regulator of the HPA axis (Fig. 2.6). Feedback inhibition by cortisol is mediated via binding to GRs in the pituitary and hypothalamus. Cortisol also acts on suprahypothalamic centers (e.g., hippocampus) to further limit the secretion of CRH [151]. Feedback inhibition in suprahypothalamic centers is mediated by cortisol binding to GRs and MRs [152].
Circadian Rhythm of Glucocorticoid Secretion
ACTH is secreted in regular pulses of varying amplitude. Peak ACTH secretion occurs in the early morning, coinciding with a rise in cortisol secretion [153]. The diurnal production of cortisol, which anticipates times of increased activity and energy demand, reflects an interplay between environmental cues (e.g., fluctuations in light intensity) and an internal circadian timekeeping system [154]. Disruption of this interplay contributes to the phenomenon of jetlag [155].
The circadian timekeeping system is composed of a central clock [the hypothalamic suprachiasmatic nucleus (SCN)] and subsidiary peripheral clocks in nearly every cell type, including adrenocortical cells [154]. The SCN is entrained to the light-dark cycle [156]. Highlighting the importance of the central clock, the diurnal glucocorticoid rhythm is altered by lesions of the SCN [157] or constant light exposure [158]. Signals from the SCN are relayed via the sympathetic nervous system to peripheral clocks in the adrenal cortex (Fig. 2.12). The rhythmic secretion of glucocorticoids from the adrenal gland is thought to synchronize peripheral circadian rhythms of tissues downstream of the SCN [155, 159].
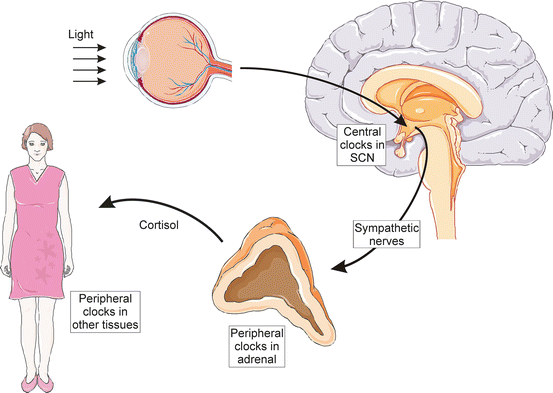
Fig. 2.12
Clock regulation of glucocorticoid secretion. Light received at the retina entrains central circadian clocks in the suprachiasmatic nucleus (SCN). Signals from the SCN are relayed via the sympathetic nervous system to peripheral clocks in the adrenal cortex. The rhythmic secretion of cortisol from the adrenal gland helps synchronize peripheral circadian rhythms of tissues downstream of the SCN. Prepared using image vectors from Servier Medical Art (www.servier.com), licensed under the Creative Commons Attribution 3.0 Unported License (http://creativecommons.org/license/by/3.0/)
The central and peripheral circadian clocks are composed of interlocking positive and negative transcriptional-translational feedback loops that oscillate with a periodicity of approximately 24 hours [154]. Genetically-engineered mice lacking of key components of the molecular clock exhibit either chronic elevation or suppression of glucocorticoid levels; rhythmic expression of certain steroidogenic genes, including Star , is altered in the adrenocortical cells of these animals [160–165]. Peripheral clocks in the adrenal cortex are thought to modulate sensitivity to ACTH, although the mechanistic basis for this effect is unclear.
MC2R and MRAPs
The trophic effects of ACTH are mediated through its plasma transmembrane G-protein-coupled receptor MC2R (Fig. 2.13). Melanocortin 2 receptor accessory proteins (MRAPs) are small membrane proteins that are essential for ACTH signaling. MRAP deficiency is one of the causes of hereditary unresponsiveness to ACTH [166]. Two types of MRAPs have been identified: MRAP1 and MRAP2. In humans, there are two distinct isoforms of MRAP1, termed MRAPα and MRAPβ, which share the same N-terminus and transmembrane domain but differ in their C-terminal domains. MRAP1 isoforms are involved in intracellular trafficking and signaling of MC2R. MRAPα has been implicated in targeting of MC2R to the plasma membrane, whereas MRAPβ appears to enhance cAMP productio n by ACTH-bound MC2R [153]. MRAP2 also is involved in trafficking of MC2R to the plasma membrane.
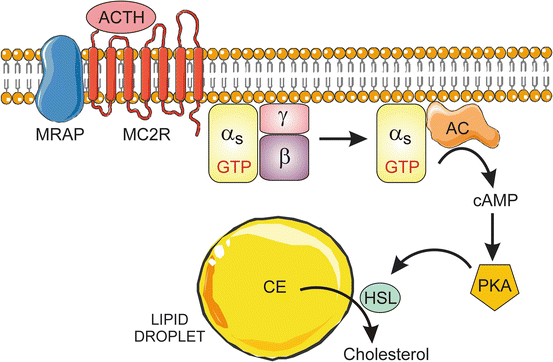
Fig. 2.13
ACTH-stimulated mobilization of cholesterol from lipid droplets. ACTH binds to its receptor MC2R. The ligand-bound receptor, in conjunction with the accessory protein MRAP, triggers the activation of AC via the heterotrimeric G-protein Gs. The resultant increase in cAMP activates PKA, leading to phosphorylation of HSL. The latter enzyme hydrolyzes CE in lipid droplets, producing free cholesterol for steroidogenesis. AC adenylate cyclase, CE cholesteryl ester, HSL hormone-sensitive lipase, MC2R melanocortin 2 receptor, MRAP melanocortin 2 receptor accessory protein, PKA protein kinase A. Prepared using image vectors from Servier Medical Art (www.servier.com), licensed under the Creative Commons Attribution 3.0 Unported License (http://creativecommons.org/license/by/3.0/)
Activation of cAMP Signaling
Binding of ACTH to MC2R causes the α-subunit of the stimulatory heterotrimeric G-protein Gs to associate with adenylate cyclase (AC), resulting in cAMP production [167]. This rise in cAMP enhances lipid synthesis, protein synthesis, and protein phosphorylation, all of which promote cholesterol trafficking to mitochondria and the production of steroid hormones [167]. Most of these effects are mediated by PKA. cAMP binds to the regulatory subunits of PKA, allowing the catalytic subunits of PKA to phosphorylate downstream effectors. For example, PKA-induced phosphorylation and activation of HSL enhances free cholesterol formation, while phosphorylation of StAR promotes transport of cholesterol from the OMM to the IMM [111]. PKA-dependent phosphorylation enhances CE uptake by enhancing the transcription and stability of SR-B1 [70]. PKA also phosphorylates transcription factors, leading to enhanced expression of steroidogenic genes such as CYP11A1 [167]. The transcription factors phosphorylated include steroidogenic factor 1 (SF1), cAMP response element-binding protein (CREB), and activator protein 1 (AP1) [150]. PKA-mediated phosphorylation of L-type Ca2+ channels activates a slow but sustained influx of Ca2+ that triggers additional signaling pathways.
Loss-of-function mutations in the protein kinase A regulatory subunit gene (PRKAR1A) cause excessive cAMP production. Such mutations underlie Carney complex, a syndrome associated with the pituitary-independent Cushing syndrome and adrenocortical neoplasia. Conditional deletion of Prkar1a in the adrenal cortex of mice leads to not only excess glucocorticoid production but also impaired apoptosis [168], mediated in part by crosstalk between the PKA and mammalian target of rapamycin (mTOR) pathways [169].
Somatic gain-of-function mutations in the PRKACA gene, which encodes the catalytic subunit of PKA, cause the ACTH-independent Cushing syndrome due to cortisol-producing adenomas [170, 171]. Germline duplications of this gene can cause bilateral adrenal hyperplasia [170].
Although the initial and most significant actions of ACTH are mediated through cAMP and the subsequent activation of PKA, there are also PKA-independent effects of cAMP, such as those mediated by Epac (the Exchange protein directly activated by cAMP) [172, 173]. One Epac isoform, Epac2, is highly expressed in the adrenal cortex and functions to activate Rap GTPases [174]. Epac proteins regulate various cellular processes via mechanisms that include modulation of gene expression and cytoskeletal rearrangements [153, 173, 174]. Treatment with an Epac-selective analogue of cAMP is sufficient to stimulate the expression of steroidogenic enzymes and cortisol secretion [175].
Phosphodiesterases
The level of intracellular cAMP is determined not only by its rate of synthesis by AC but also its rate of degradation by phosphodiesterases (PDEs) [176]. In mammals there are 11 families of PDEs, each with distinctive properties [177]. In mice, treatment with a PDE-selective inhibitor increases phosphorylation of HSL and basal corticosterone secretion [178]. Loss-of-function mutations in PDE8B or PDE11A have been linked to bilateral adrenal hyperplasia and Cushing syndrome in humans [176, 178].
Hydrolysis of cAMP by PDEs produces AMP, which in turn stimulates the AMP-activated protein kinase (AMPK) [179]. Activated AMPK represses the expression of transcription factors known to stimulate steroidogenesis (e.g., NUR77 and cJUN) and activates the expression of repressors of steroidogenesis (e.g., DAX1 and cFOS) [180] (Fig. 2.14).
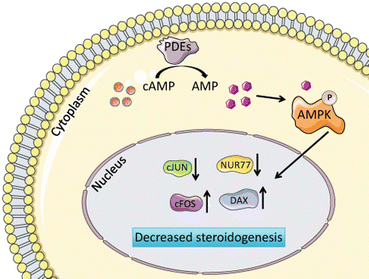
Fig. 2.14
Termination of cAMP signaling. Hydrolysis of cAMP by PDEs produces AMP, which in turn stimulates AMPK. Activated AMPK represses the expression of transcription factors known to stimulate steroidogenesis (e.g., NUR77 and cJUN) and activates the expression of repressors of steroidogenesis (e.g., DAX1 and cFOS). Prepared using image vectors from Servier Medical Art (www.servier.com), licensed under the Creative Commons Attribution 3.0 Unported License (http://creativecommons.org/license/by/3.0/)
Interplay of cAMP Signaling and Arachidonic Acid Metabolism
ACTH stimulation induces the release of arachidonic acid (AA) from phospholipid stores, and this fatty acid and its metabolites have been shown to enhance steroidogenesis [181, 182]. Intracellular levels of free AA are tightly regulated by not only phospholipases, such as phospholipase A2 (PLA2), but also by acyl-CoA synthetases and thioesterases. Most acyl-CoA synthetases exhibit a broad specificity with respect to fatty acid substrate, but acyl-CoA synthetase 4 (ACSL4) prefers AA as a substrate [183, 184]. Originally characterized as an activity in platelets [185], ACSL4 is highly expressed in steroidogenic tissues including the adrenal gland and gonads [183, 186]. The expression of Acsl4 mRNA is upregulated through a cAMP-dependent pathway , and pharmacological inhibition of ACSL4 reduces steroidogenesis in adrenocortical cell lines [187]. The hormonal regulation of steroid synthesis requires the concerted action of ACS4 and a second enzyme, mitochondrial acyl-CoA thioesterase 2 (ACOT2), which cleaves arachidonoyl-CoA into its component parts. These two enzymes constitute an AA generation/export system, which releases AA in mitochondria in response to ACTH stimulation [188]. AA is then metabolized into lipoxygenated or epoxygenated products that induce the expression of StAR [182, 187, 188].
Other Signaling Pathways Activated by ACTH
ACTH triggers a transient increase in total protein tyrosine phosphatase (PTP) activity in zF cells and a concomitant decrease in the phosphotyrosine level of several proteins [189]. Treatment with PTP inhibitors reduces hormone-induced stimulation of steroidogenesis [153], and PTPs have been implicated in the regulation of StAR induction and cholesterol transport to the IMM [189]. One particular PTP, PTPN11, has been shown to regulate the expression of ACSL4 [190].
MAPKs have also been implicated in the action of ACTH [24]. This kinase family comprises the extracellular signal-related kinases ERK1/2 (p42/p44mapk), the p38 MAPKs, and the p54 stress-activated JNK protein kinases [191]. MAPK targets in steroidogenic cells include HSL [76] and CYP17A1 (see later).
JAK/STAT (Janus kinase/signal transducer and activator of transcription) signaling is also activated by ACTH. Pharmacological or siRNA-mediated inhibition of JAK2 impairs ACTH-induced steroidogenesis [192]. Nuclear JAK2 regulates the amount of active CREB through tyrosine phosphorylation and prevention of proteasomal degradation, which in turn leads to transcriptional upregulation of StAR [192].
ACTH-Independent Mechanisms Regulating Cortisol Secretion
Although ACTH is the principal stimulus for glucocorticoid secretion , there are several ACTH-independent mechanisms that regulate cortisol release from the adrenal cortex. A variety of neuropeptides, neurotransmitters, and cytokines bind receptors on the surface of zF cells and modulate glucocorticoid secretion [193]. Additionally, factors secreted by endothelial cells and adipocytes influence the secretion of adrenal steroids [194, 195]. The close anatomical relationship among adrenocortical cells, medullary chromaffin cells, and nerve endings facilitates paracrine modulation of adrenal secretion of glucocorticoids [193].
Regulation of Aldosterone Secretion
The zG is optimized for the synthesis of aldosterone. Cells in this zone express CYP11B2 (aldosterone synthase) but not CYP17A1, the enzyme that directs steroid substrates toward cortisol and androgen synthesis. The principal controllers of aldosterone production are Ang II and extracellular K+, although ACTH also plays a role [17]. Ang II and extracellular K+ act mainly by generating a cytosolic Ca2+ signal [196]. The signaling pathways and effectors employed include phospholipase C (PLC), inositol 1,4,5-trisphosphate (IP3), Ca2+/calmodulin-dependent protein kinases (CaMKs), diacylglycerol (DAG), PKC, MAPKs, tyrosine kinases, and PKA. Stimulation of these signaling pathways leads to the direct activation of several transcription factors. This, in turn, increases the expression of steroidogenic genes, including CYP11B2.
ACTH Signaling in the zG
In addition to regulating cortisol secretion in zF cells, ACTH can induce aldosterone production in the zG. As in the zF, ACTH acts on zG cells by binding to its receptor, MCR2, and activating AC via Gs [17]. This leads to increased cAMP/PKA signaling, which facilitates the movement of cholesterol from the OMM to the IMM and activates transcription of key steroidogenic genes. Binding of ACTH to its receptor also impacts the electrical properties of zG cells [24].
Ang II Signaling
The zG controls extracellular fluid volume and salt balance as part of the renin-angiotensin-aldosterone system [17, 23]. Renin is a protease secreted by the juxtaglomerular apparatus of the kidney in response to extracellular fluid depletion, low sodium concentrations, or hypotension. Renin cleaves angiotensinogen, a glycoprotein constitutively secreted into the serum by the liver, yielding the decapeptide angiotensin I (Ang I). Angiotensin-converting enzyme (ACE), a protein expressed on the surface of pulmonary and renal endothelial cells, subsequently converts Ang I into to the vasoactive octapeptide Ang II.
Related posts:
 Pheochromocytomas and Paragangliomas: Genetics and Pathophysiology
Pheochromocytomas and Paragangliomas: Genetics and Pathophysiology
 Adrenal Zonation and Development
Adrenal Zonation and Development
 Diagnosis and Management of Adrenal Insufficiency
Pheochromocytomas and Paragangliomas: Genetics and Pathophysiology
Diagnosis and Management of Adrenal Insufficiency
Pheochromocytomas and Paragangliomas: Genetics and Pathophysiology
 Genetics and Pathophysiology of Congenital Adrenal Hyperplasia
Genetics and Pathophysiology of Congenital Adrenal Hyperplasia
 Adrenal Cushing’s Syndrome: Updates on Overt and Mild Hypercortisolism
Adrenal Cushing’s Syndrome: Updates on Overt and Mild Hypercortisolism
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


