Fig. 18.1
Chemical structures of cytidine and the cytidine analogs azaC (azacytidine) and DAC (aza-2′-deoxycytidine). The modifications of nitrogen in the base are depicted with red color. The red arrow highlights the deficiency of the deoxy group on the ribose molecule of DAC compared to cytidine and AzaC
Naturally, deoxycytidine is incorporated into the DNA chain during replication and forms a bridge with the corresponding guanine base. Sequences in the DNA with deoxycytidine–deoxyguanosine (CpG) dinucleotides occur at a high frequency, i.e., an observed-to-expected CpG ratio that is greater than 60%, are referred to as CpG islands [5]. These CpG islands are predominantly located in the promoter region of genes. Gene promoters of human genome consist 72% of high CpG content [6]. Deoxycytidine within CpG dinucleotides can be methylated to form 5-methyldeoxycytidine. By DNA methylation of a gene promoter, a gene can be silenced. In eukaryotes DNA methylation is essential for development, genomic imprinting, and X chromosome inactivation. DNA methylation is also implicated in cancer development and progression by silencing of tumor suppressor genes upon promoter methylation. DNA methylation is a reversible process that can be exploited for cancer treatment. Since AzaC and DAC interfere with the process of methylation, these drugs function as epigenetic modulators [3] and can reverse the cancer phenotype. Genes silenced by promoter CpG island hypermethylation in cancers, that are reported to be reactivated after treatment with AzaC or DAC, are summarized elsewhere [7].
Methylation of deoxycytidines, also called cytosine methylation, is carried out by DNA methyltransferases. The methylation pattern is read and copied to a newly synthesized DNA strand by the maintenance methyltransferase DNMT1, which has affinity for hemi-methylated DNA strands [8]. A novel (not earlier present on the DNA) methyl group can be added by DNMT3A or DNMT3B, which are de novo DNA methyltransferases with prevalence for unmethylated DNA [9, 10].
DNMTs use S-adenosyl methionine as the methyl donor (AdoMet). The catalytic mechanism of DNA methyltransferase involves a covalent bond formation between a cysteine residue in the active site of the enzyme and carbon 6 of cytosine in DNA. Thereby, an increase in the flow of electrons to carbon 5 is initiated generating an attack on AdoMet. Extraction of a proton from carbon 5 subsequently follows β-elimination, and a double bond between carbon 5 and carbon 6 is formed. As a result the enzyme and S-adenosyl-homocysteine (AdoHcy) are released, leaving methylated cytosine in the DNA. Cytosine is suitable as methyl-binding moiety because the cytosine is a flexible base, which flips out of the DNA prior to receiving the methyl group. This phenomenon has been described as base-flipping mechanism in various studies [11–13].
The effectivity of epigenetic modifiers as anti-cancer agents is depending on several cellular processes such as uptake and metabolism of the drugs and cell proliferation. Moreover, several effects can be anticipated both on DNA and RNA. To optimize the epigenetic treatment for different cell types, currently several novel (antimetabolite) drugs have been developed, which are described in detail in this review.
18.2 Uptake and Metabolism of AzaC and DAC
In order for nucleoside analogs, such as AzaC and DAC, to function, they need to be internalized into the cells. The nucleoside transporter family SLC29A gene codes for the equilibrative nucleoside transporter (ENTs) proteins and the SLC28A gene for concentrative nucleoside transporters proteins (CNTs). These transporters are responsible for intracellular transport and have been related to nucleoside analog sensitivity in leukemia [14–17]. AzaC is a substrate for hCNT1 and hCNT3. Transport of AzaC into the cell is therefore mediated by these cell membrane transporters (Fig. 18.2) [18, 19]. Moreover, the human ENT is also involved in the transport of AzaC and DAC [20]. Transporter deficiency can lead to resistance to these drugs.
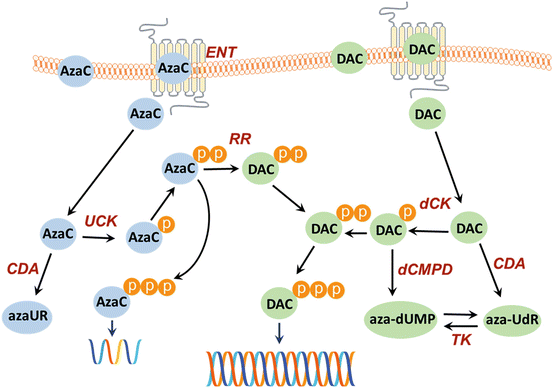
Fig. 18.2
Uptake and metabolism of AzaC and DAC. Compounds enter the cell by diffusion or membrane transporter-mediated influx (human equilibrative nucleoside transporter, hENT). Human concentrative nucleoside transporters (hCNTs) can also play a role in the uptake. The drugs can be inactivated by deamination, catalyzed by cytidine deaminase (CDA). In the cell the metabolic inactive AzaC and DAC are activated by means of phosphorylation, azaC by uridine–cytidine kinase (UCK), and DAC by deoxycytidine kinase (dCK). DAC-MP can also be inactivated by dCMP-deaminase (dCMPD) to aza-dUMP. The deaminated DAC, aza-UdR, is a substrate for thymidine kinase (TK), which catalyzes the transfer of aza-UdR to aza-dUMP. AzaC-DP is reduced by ribonucleotide reductase (RR) to its deoxy-form, aza-2′-deoxycytidine diphosphate (DAC-DP). AzaC-TP is incorporated into RNA; DAC-TP is incorporated into DNA. Incorporation of DAC-TP into DNA leads to hypo-methylation since this will inhibit DNA methyltransferase
After the translocation of the nucleosides into the cells, the prodrugs have to be activated in order to exert their antimetabolite activity [21]. The activation is carried out by phosphorylation. The phosphorylation cycle of AzaC generates AzaC-MP, AzaC-DP, and AzaC-TP (Fig. 18.2). When AzaC is phosphorylated to the AzaC-TP form, it can be incorporated into the RNA. For incorporation into the DNA, the ribose of AzaC-DP needs to be reduced to deoxyribose, which relies on ribonucleotide reductase (RR) [22]. The activating enzyme for AzaC is the uridine–cytidine kinase (UCK) [23], while for DAC deoxycytidine kinase is responsible for activation/phosphorylation of the drug [24]. A known mechanism of deactivation of cytidine analogs is deamination. Both AzaC and DAC are substrates for cytidine deaminase and can be deaminated within the cells, while aza-dCMP can also be deaminated by deoxycytidylate deaminase (Fig. 18.2) [22, 25, 26].
The epigenetic modifying drugs interfere with the activity of DNA-methylating enzymes (DNA methyltransferases), which are often deregulated in cancer.
18.3 The Maintenance DNA Methyltransferase 1 Protein
The maintenance DNA methyltransferase DNMT1 is ubiquitously expressed and has several interacting proteins that are listed elsewhere [27]. DNMT1 is composed of different domains that can be given a functional importance. The N-terminal site of DNMT1 is a regulatory region and consists of proliferating cell nuclear antigen (PCNA) binding domain, (PBD), a heterochromatin targeting sequence (TS), a CXXC-type zinc finger domain, and two bromo-adjacent homology domains (BAH1 and BAH2). The C terminus has the catalytic activity. PCNA targets DNMT1 to the DNA replication site and DNA repair sites in order to restore the DNA methylation pattern [28–31]. It is important to note that methylation is enhanced when the replication machinery is involved, but the assembly of replication fork is not necessary to maintain DNA methylation [32, 33]. The heterochromatin targeting domain was shown to recruit DNMT1 to heterochromatin independent of DNA replication [30]. The CXXC-type zinc finger domain interacts with DNA and ensures that the enzyme is in an auto-inhibitory state in the presence of unmethylated DNA [34]. The N terminus and C terminus of the protein are connected by lysyl–glysyl dipeptide repeats linker ((KG)7) [35]. The function of the C terminus is the catalytic activity of the enzyme, which uses AdoMet as a donor. Several post-transcriptional modifications have been reported to be important for stability and activity of DNMT1. The decay of the protein is proteasome mediated whereby the protein is tagged by ubiquitination [36]. The E3 ligase activity containing protein UHRF1 can ubiquitinate DNMT1 [37]. Phosphorylation and methylation of DNMT1 are reported to increase the stability of the protein [38–41]. In addition, SUMOylation increases in DNMT1 activity [42]. A critical role for DNMT1 in development was demonstrated in animal models; DNMT1 null mice die after gastrulation [43]. DNMT1 overexpression and/or hyperactivity has been reported in various tumors [44–46].
18.4 De Novo Methyltransferases in Health and Disease
In comparison to DNMT1, de novo DNA methyltransferases are tissue-specific [44] smaller proteins. DNMT3A is comprised of 912 amino acids, while DNMT3B is even smaller and is comprised of 853 amino acids. DNMT3A persists in three isoforms. The canonical isoform is 912 amino acids long and the others 723 and 166 amino acids, respectively [10]. The isoform variants are generated by means of alternative splicing. In non-diseased conditions a cell uses alternative splicing to ensure proteomic diversity [47]. Functionally different proteins are then formed from one gene [48]. Alternative splicing is deregulated during cancer development and progression and can cause drug resistance [49, 50]. Interestingly, an alternative spliced variant of DNMT3B was found in tumors, which has an altered functionality [51]. DNMT3A acts in complex with DNMT3L [52, 53], which is inactive on its own [54]. DNMT3L is a stimulatory cofactor of the DNMT3A and DNMT3B enzymes [55]. The DNMT3A gene is frequently mutated in acute myeloid leukemia (AML) [56, 57] and chronic myelomonocytic leukemia (CMML) [58], and recently a condition named Tatton–Brown syndrome was reported with mutations in DNMT3A gene [59]. On the other hand, mutations in DNMT3B gene cause immunodeficiency–centromeric instability–facial anomalies syndrome 1 (ICF1) [60–64]. For this gene eight different isoforms have been reported. DNMT3A and DNMT3B enzymes are thought to have preference for target sites [65, 66].
18.5 The Mechanism of DNMT1 Downregulation Upon AzaC and DAC Treatment and Beyond
18.5.1 Replication-Based DNMT1 Downregulation
AzaC and DAC have an effect on the DNA methylation pattern. Therefore, these compounds are known as epigenetic modulators. The mechanism of epigenetic modulation involves downregulation of DNA methyltransferases (DNMTs). DAC is able to downregulate DNMT1, DNMT3A, and DNMT3B. Because of the implication of DNMT1 in tumor suppressor gene promoter hypermethylation, interest in DNMT1 downregulating agents has been increasing. Early studies indicated downregulation of DNMT1 by a trapping mechanism. Thereby aza-dCTP is incorporated into the DNA, and a covalent binding is formed between the false cytosine and DNMT1 (Fig. 18.3). Subsequently, the methyltransferase is degraded [67–69]. Mass spectrometry analysis of DNA isolated from DAC-treated cells also revealed an open-ringed aza-cytosine lacking its carbon 6. It was proposed that the enzyme can still be bound to the base in the open-ring format [70].
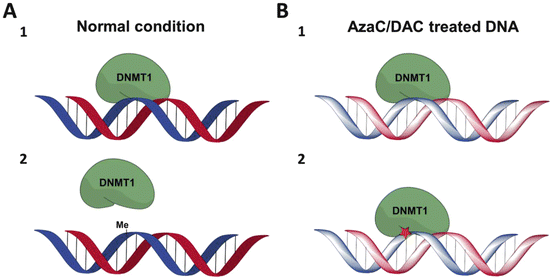
Fig. 18.3
Simplified overview of DNMT1 trapping by AzaC and DAC. a Normal condition of DNA methylation by DNMT1. 1 DNMT1 recognizes hemi-methylated DNA and interacts with it, in order to methylate the cytosine base. 2 The cytosine methylation has occurred; DNMT1 release takes place. b DNA methylation by DNMT1 after treatment with AzaC and DAC. 1 DNMT1 recognizes hemi-methylated DNA and interacts with it. 2 Because of incorporated DAC, DNMT1 forms covalent binding with DNA; the enzyme is not released instantly
18.5.2 Ubiquitination of DNMT1 Upon DAC Exposure
Ubiquitination plays a role in DAC-induced DNMT1 downregulation [71]. Cellular protein degradation is mediated but not limited by ubiquitination, which tags protein for proteasomal degradation. Ubiquitin itself is a polypeptide of 76 amino acids. The cascade of polyubiquitination starts by the ubiquitin-activating enzyme E1 [72]. E1 catalyzes in an ATP-dependent manner the covalent binding of ubiquitin to the cysteine residue of the E1 active site. Then the activated ubiquitin is transferred to ubiquitin-conjugating enzymes, E2. E2 transfers ubiquitin to the third enzyme, ubiquitin protein ligase, E3. Glycine (ubiquitin)–lysine (protein residue) bound is thereby formed. It is generally believed that lysine 48 polyubiquitin chain is a signal for proteasomal degradation [73–76]. Therefore, lysine 48 is a known tag for proteins to degrade. Evidence for ubiquitin-associated degradation of DNMT1 upon DAC exposure was demonstrated, in that upon inhibition of proteasomal activity using the compound MG 132, no DNMT1 downregulation was seen in DAC-treated cells [71, 77].
18.5.3 The Effect of AzaC on RNA
One of the additional characteristics of AzaC is the ability to be incorporated into the RNA. Next to affecting RNA synthesis, incorporation of AzaC into the tRNA was shown to inhibit tRNA methyltransferases, which results in interference with tRNA methylation and processing leading to defective acceptor function of tRNA [78–80]. In addition, incorporation of AzaC into the nucleolar RNA was shown; ribosomal precursor RNA containing AzaC was suggested to have an altered structure [81]. Since methylation is of great importance for ribosomal RNA processing, incorporation of AzaC into RNA will affect protein synthesis as well [82–85]. Interestingly, the deamination products of AzaC and DAC, azauridine (when sequentially phosphorylated to aza-UMP and aza-UDP, which will be reduced to aza-dUDP and dephosphorylated to aza-dUMP) and azadeoxyuridine (when phosphorylated directly to aza-dUMP) interfere with de novo thymidylate synthesis, since aza-dUMP is a poor inhibitor of thymidylate synthase, which may enhance cytotoxic effect of both compounds [86]. Aza-dUMP can also be formed by deamination of aza-dCMP.
18.6 Novel DNMT Inhibitors
Because of the chemical instability of both azacytidine and DAC, prodrugs and novel analogs of both compounds have been designed. Moreover, several compounds were also designed to offer the possibility for oral administration. Zebularine [87–89], SGI-110 (oligonucleotide consisting of DAC linked through a phosphodiester bond to the endogenous nucleoside deoxyguanosine) [90, 91], TAC (2′,3′,5′-triacetyl-5-azacytidine) [92], CP-4200 (elaidic acid ester derivative of AzaC) [93], T-dCyd (4′-thio-2′-deoxycytidine) [94], and Aza-T-dCyd (5-aza-4′-thio-2′-deoxycytidine) [94] are examples of second-generation cytidine nucleoside analogs. The structure and update on clinical trials of these compounds are listed in Table 18.1.
Table 18.1
Overview of the next generation DNMTs downregulators
Chemical structure/compound name | Type of cancer | Status/administration | References |
|---|---|---|---|
Prodrugs | |||
 SGI-110 | AML | Clinical studies | [102] |
MDS | Phase 1, 2, and 3 | [90] | |
CMML | s.c. injection | ||
Advanced HCC | |||
Metastatic melanoma | |||
Metastatic colorectal cancer | |||
Ovarian cancer | |||
Germ cell tumor | |||
 TAC | In vivo model: | Preclinical studies | [92] |
Antileukemic activity in L1210 BDF1 female mice | Oral administration | ||
i.p. injection | |||
 CP-4200 | In vivo model: | Preclinical studies i.p/i.v. injection | [93] |
ALL SCID6 diabedic/severe immunodeficient female mice | |||
Analogs | |||
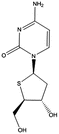 T-dCyd | Advanced solid tumors | Clinical studies Phase 1 | [94] |
Oral administration | |||
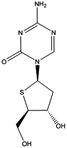 Aza-T-dCyd | In vivo model: | Preclinical studies | [94] |
Antitumor effect in NCI-H23 lung tumor implanted nude mice | i.p. injection | ||
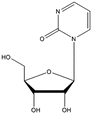 Zebularine | In vivo models: | Preclinical studies | [103] |
Effective in genetically engineerd MMTV-PyMT mammary tumor mice | Oral administration | [104] | |
Inhibition of adenoma formation in C57/BL6 female Apc Min/+ mice | |||
Activity in Panc-89 injected NMRI mice | i.p. injection | [105] | |
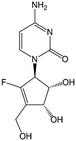 RX-3117 | Advanced solid tumors | Clinical studies | [106] |
Phase 1 and 2 | |||
Oral administration | |||
The uptake of the cytidine analog zebularine into the cell is mediated by hCNT1, hCNT3, and hENT2 [95]. When phosphorylated the compound has a potent inhibitory effect on DNMT1 protein, and to a lesser extent, it inhibits also DNMT3A and DNMT3B [96]. Zebularine exerts its action by trapping DNMT1 and preventing its turnover [97]. In addition to downregulation of DNMTs, zebularine was shown to inhibit CDA by an adduct formation with the active site of CDA [98]. Zebularine has no clinical trial records registered yet. On the other hand, the dinucleoside SGI-110 is designed to take care of deamination and instability of DAC [99] and showed potent activity in malignant xenograft models [91]. Currently, there are 13 clinical trials in phases 1, 2, and 3, evaluating this compound alone or in combination with other compounds commonly used in cancer treatment. The compound is being tested in various malignancies.
The compound TAC ,which is a prodrug of AzaC, performed better in terms of oral bioavailability, stability, and solubility as it was initially aimed for clinical studies [92]. However, to the best of our knowledge, the drug did not enter a clinical trial yet. Interestingly, T-dCyd is activated by dCK and incorporated into the DNA without DNA synthesis inhibition and downregulation of DNMT1. The modification in this compound enabled poor deamination, and it can be administered orally. The compound recently entered phase 1 clinical trials and is being studied in advanced solid tumors [100]. Both, T-dCyd and Aza-T-dCyd had in vivo efficacy and are thought to have limited ability to inhibit thymidylate synthase. However, both compounds remain an experimental drug [94].
Cellular uptake of cytidine analogs and clinical efficacy, however, rely often on membrane transporters. AzaC uptake is transporter mediated and was shown to have limited drug uptake. To overcome this limitation, CP-4200 was designed. CP-4200 uptake into the cytosol was independent of nucleoside transporters. In the cell the elaidic acid chain is cleaved off, and azaC is released. Therefore, the mechanism of action is similar to AzaC by DNMT downregulation by trapping of the enzyme [93]. CP-4200 is an interesting experimental drug for its potential to overcome transporter-mediated resistance to AzaC [101].
A chemically different but functionally similar DNMT downregulator is the cytidine analog fluorocyclopentenylcytosine (RX-3117) [106–109]. Initial studies on its mechanism of action and metabolism have been published recently. Uptake of RX-3117 is mediated by hENT1 and its phosphorylation by UCK2 [107]. UCK2 is overexpressed in tumors, which makes RX-3117 specific for cancer cells and possibly will have a low systemic toxicity [107]. Furthermore, it was shown that RX-3117, contrary to AzaC and DAC, is not deaminated by cytidine deaminase (CDA) and that RX-3117 causes both inhibition of DNA and RNA synthesis, although the inhibition of the former is more pronounced. By doing so, RX-3117 induces DNA double-strand break damage [110]. Interestingly, RX-3117 also targets DNA methyltransferase (DNMT). In two studies a decrease in DNMT1 expression was found in cell lines treated with RX-3117, while this was not the case for DNMT3A [106]. RX-3117 is able to reactivate functional proteins, such as the proton-coupled folate transporter, and repair enzymes, such as O-6-methylguanine DNA methyltransferase and tumor suppressor genes [109]. The mechanism of DNMT1 downregulation by RX-3117, however, is unknown yet. Together these data indicate that RX-3117 might be an effective demethylating agent, comparable to decitabine (Aza-CdR) and azacytidine (Aza-CR) [21].
Since the nucleoside cytidine analogs showed dose-limiting toxicity, non-nucleoside DNMT1 inhibitors have also been developed. The design of such inhibitors is based on computer modeling of catalytic domain of the protein. The non-nucleoside DNMT1 downregulator, N-phthalyl-L-tryptophan (RG108) [111], has shown a potent demethylation effect in vitro [112–114]. Naturally occurring non-nucleoside analogs also exist: tea polyphenol, epigallocatechin- 3gallate (EGCG), genistein, nanomycin A, psammaplin A, and the laccaic acid A. The mechanism of DNMT downregulation of these non-nucleoside compounds is not described yet. These compounds also differ considerably in chemical structure [111].
18.7 Conclusions
Major efforts have been put into the development of novel DNA-demethylating agents. Increasing knowledge from existing drugs permits us to learn about their mechanism of action and enables to develop superior derivatives. A compound can be modified to overcome very well-known existing resistance mechanism or to increase its efficacy. The availability of improved techniques for structural studies on target proteins will likely result in the development of more specific drugs in the future. Application of cancer biology led to the development of a unique novel DNMT1 downregulator, RX-3117. This drug combines a cancer-specific key enzyme necessary for the activation of metabolic inactive drug, with DNMT1 as the ultimate target, which makes this drug a promising anticancer treatment.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


