Fig. 9.1
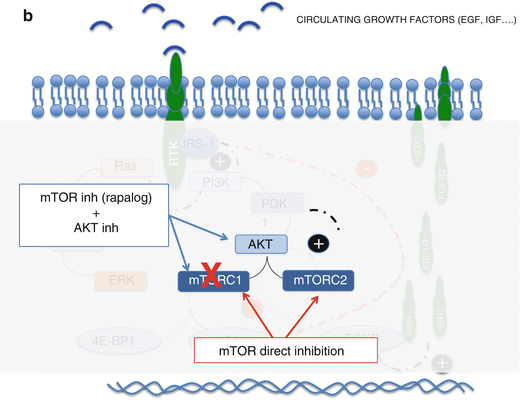
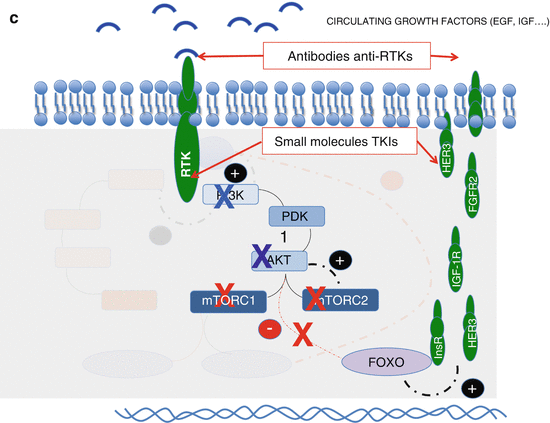
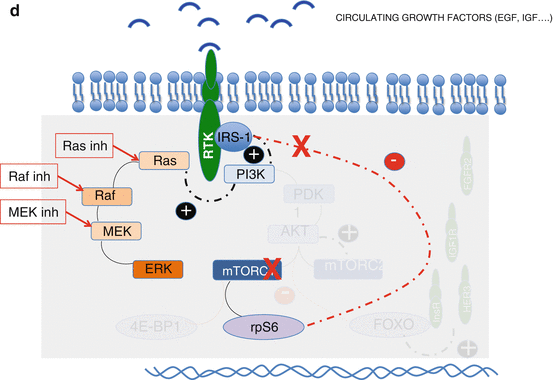
(a) Feedback loops of the PI3K/Akt/mTOR pathway. Red line means inhibition and black activation; in blue: PI3k/Akt/mTOR pathway; in brown: MAPK pathway. (b) Two ways of blocking the AKT activation by mTORC2. (c) Blockade of RTKs overexpressed due to dimished activiy of AKT (in case of AKT, PI3K or direct mTOR inhibition). (d) Blockade of the MAPK pathway that is activated by PI3K when dimished activity of pS6 derepresees IRS-1
, three major feedback loops have been described that seem to affect mTOR inhibitors efficacy:
Akt activation by TORC2. mTOR has been shown to be the key component of two different complexes named TORC1 and TORC2. The second is insensitive to rapalogs and is known to directly phosphorylate Akt [8]. Thus, TORC1 inhibition alone cannot control this escape route.
Enhanced expression of receptors tyrosine kinases (RTKs). Akt represses the activity of a group of transcription factors named FOXO that regulates the expression of several RTKs including the fibroblast growth factor receptor (FGFR), insulin receptor (insR), insulin growth factor receptor (IGF-1R), and HER3 among others. Therefore, any decrease in Akt activity will enhance the expression of these receptors that are known to use the PI3K/AKT/mTOR and the MAP kinase pathways as second messengers.
MAPK pathway activation by PI3K and IRS-1. PI3K does not only stimulate PDK1 and the downstream factors within the PI3K/AKT/mTOR pathway but also the MAPK pathway (Fig. 9.1a). This dual activation is a key element of the cross-talk between both pathways and a major mechanism of resistance to mTOR inhibitors.
Insulin receptor substrate (IRS-1) is a good example of how this resistance works. IRS-1 is known to activate PI3K, and, in physiological conditions, its activity and expression are repressed by pS6 [9]. When mTOR inhibition leads to pS6 decrease, IRS-1 is released leading to PI3k activation and MAPK stimulation.
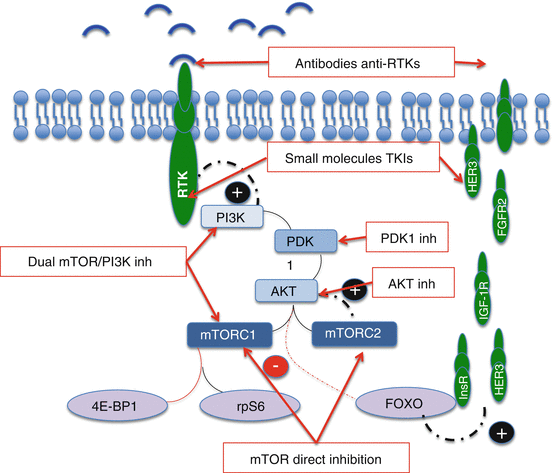
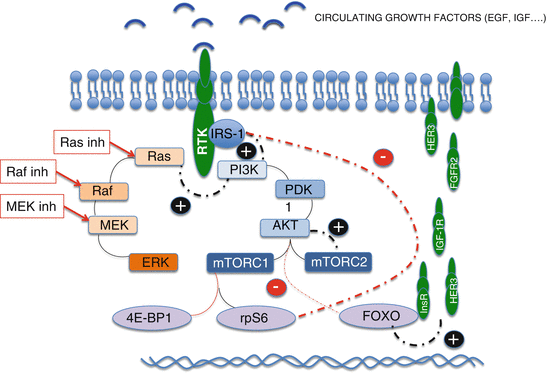
Fig. 9.2
Vertical inhibition of the PI3K/Akt/mTOR pathway. Red line means inhibition and black activation; in blue: PI3k/Akt/mTOR pathway; in brown: MAPK pathway
Fig. 9.3
Horizontal inhibition of the PI3K/Akt/mTOR pathway and MAPK. Red line means inhibition and black activation; in blue: PI3k/Akt/mTOR pathway; in brown: MAPK pathway
In conclusion, when developing combinations with mTOR inhibitors, we can follow three different strategies:
In next sections, we will review the theoretical advantages and pitfalls of such strategies and the so far published clinical experiences. Finally, we will provide insights regarding the most promising lines of investigation.
9.3 Vertical Inhibition Strategies
9.3.1 Enhancement of mTOR Complex (mTORC) Inhibition
9.3.1.1 Metformin
Probably one of the most intriguing molecules in oncology over the last years has been metformin, a compound first synthesized in the 1920s. It is widely used as antidiabetic, and compelling literature has demonstrated a lower incidence of cancer in diabetic patients taking this drug [10–12].
In cellular models, metformin has demonstrated to induce AMPK formation that ultimately stimulates the tuberous sclerosis complex 1 (TSC1), an mTORC1 inhibitor, and phosphorylates Raptor, a component of mTORC1, leading to its inactivation [13–15].
Thus, combining metformin with rapalogs would produce a double disassembly of mTORC1 and the activation of its key inhibitor, TSC1. Additionally, from a clinical point of view, adding an antidiabetic drug to rapalogs could help to control or even prevent the development of hyperglycemia, a class effect of these compounds.
Despite its theoretical interest, only one phase I clinical trial has already been communicated combining temsirolimus and metformin [16]. Relevant toxicity was observed when standard doses of both drugs were combined. Some ongoing clinical trials will provide more data about the efficacy of this combination.
9.3.1.2 Farnesyltransferases Inhibitors
Rheb (RAS homolog enriched in brain) is an activator of mTORC1 that depends on farnesyltransferation to become active. Thus, farnesyltransferases inhibitors could potentially enhance mTORC1 blockade [17].
However, disappointing results have been achieved with this class of drugs, and no combination with rapalogs has been tested. In fact, their clinical development is questioned [18].
9.3.1.3 Direct mTOR Inhibition
Rapalogs inhibit mTOR function by binding a component of mTORC1: the FK506 binding protein 12 (FKBP12). Thus, they do not directly interact with mTOR but induce disassembly of mTORC1 complex repressing its activity.
Since FKBP12 is not present in mTORC2, none of these compounds will inhibit this complex that is known to activate AKT through a feedback loop (Fig. 9.1a). This is considered a key fact regarding the limited results of rapalogs in the clinic.
The development of small molecules that directly inhibit the catalytic site of mTOR would block both complexes (mTORC1 and 2) avoiding such feedback loop (Fig. 9.1b). This is an evolving field with several compounds under evaluation, most in preclinical studies or early phase I trials (Table 9.1).
Table 9.1
mTORC1/2 inhibitors under clinical development
Communicated clinical trials | Comments | Tumors treated | |
|---|---|---|---|
OSI-027 | Phase I [19] | Completed by February 2013 (results awaited) | Solid malignances |
MLN-0128 (INK-0128) | Phase I | One trial completed by March 2013 (results awaited). Two ongoing | Multiple myeloma, Waldenstrom, and solid malignancies |
AZD8055 | Phase I [20] | One trial already Communicated with liver toxicity as main AE | Liver cancer, gliomas, and other solid malignances |
AZD2014 | Phase I [21] and II | Phase I combined with fulvestrant is under development; a comparative phase II trial vs everolimus in kidney cancer is ongoing | Breast and kidney cancer and other solid malignancies |
Regarding the combinations of these compounds, the rationale is quite similar to rapalogs and will be discussed in depth in Sect. 9.4. For instance, preclinical models have demonstrated synergistic activity of OSI-027 (a direct mTOR inhibitor) with epidermal growth factor receptor (EGFR) and Her-2 inhibitors [22, 23]. AZD8055 has been combined in vitro with different MEK inhibitors achieving promising results, and one trial is currently evaluating the use of concomitant AZD 2014 plus fulvestrant, resembling the studies performed with everolimus plus aromatase inhibitors in breast cancer [24, 25]. The results of the ongoing clinical trials are eagerly awaited.
9.3.2 Upstream mTOR Inhibition
9.3.2.1 PI3K Inhibitors
PI3k inhibitors are another area of intensive research. Though combining these compounds with mTOR inhibitors could lead to a more potent repression of the PI3k/Akt/mTOR pathway, the development of dual PI3K and mTOR inhibitors has emerged as the preferred option. Thus, such combinations will no longer be studied.
However, PI3K inhibitors are being extensively studied in a number of schedules with other drugs (Table 9.2).
Table 9.2
PI3K inhibitors under clinical development
Communicated clinical trials | Target | Combinations under development | Tumors treated | |
|---|---|---|---|---|
PX-866 | Pan PI3K | Docetaxel Cetuximab Vemurafenib | Prostate cancer, melanoma, glioblastoma, and other solid malignances | |
GDC-0941 | PI3K | MEKi Anti-EGFR Trastuzumab chemotherapy | Non-Hodgkin lymphoma, multiple myeloma, breast cancer, and other solid malignances | |
BYL719 | Phase I [33] and II | PIK3alpha | Hormone therapy Anti-EGFR FGFRi MEKi HSPi Imatinib BRAFi Antiangiogenics | CRC, NSCLC, esophageal and pancreatic cancer, and other solid malignances |
Idelalisib (CAL-101 [GS-1101]) | p110-delta PI3K | Rituximab Bendamustine Bortezomib Ofatumumab | Indolent B cell NHL, mantle cell lymphoma, and CLL | |
XL-147 (SAR245408) | Pan PI3K | Erlotinib Trastuzumab Hormone therapy Chemotherapy | Breast and endometrial cancer, glioblastoma, lymphoma, and other solid malignances | |
Buparlisib (BKM120) | Phase I [43] and II | Pan PI3K | Bevacizumab Vemurafenib MEKi Hormone therapy Chemotherapy Imatinib PARPi Anti-EGFR mTORi Anti-Her2 | Advanced leukemias, CRC, NSCLC, RCC, breast, prostate, endometrial, and squamous head and neck carcinoma, GIST, melanoma, and other solid malignancies |
BAY 80–6946 | PIK3 | MEKi Chemotherapy | Non-Hodgkin lymphoma and solid malignances | |
GSK2636771 | Phase I/II | PIK3 | Solid malignances |
9.3.2.2 Dual PI3K/mTOR Inhibitors
mTOR and PI3K share a high degree of sequence homology in their catalytic sites. This similarity has led to the development of compounds able to inhibit simultaneously both enzymes.
There are several advantages of combining the inhibition of the two enzymes in just one drug. First, dose finding studies require less number of patients, and results are more reliable than combining an mTOR inhibitor plus a PI3K inhibitor. Second, expected toxicity should be lower using only one compound.
Regarding efficacy, the most striking interest of these drugs is their ability to avoid two relevant feedback loops that are known to be detrimental when using rapalogs:
As they inhibit directly mTOR, both complexes (mTORC1 and 2) are blocked preventing a compensatory Akt activation.
Direct PI3K inhibition would also avoid an activation of the MEK pathway through the pS6-PI3K-Ras feedback loop (Fig. 9.1a).
A summary of drugs under development is provided in Table 9.3.
Table 9.3
Dual PI3K/mTOR inhibitors under clinical development
Communicated clinical trials | Target | Combinations under development | Tumors treated | |
|---|---|---|---|---|
NVP-BEZ235 | Phase II | PI3K/mTOR | Everolimus Trastuzumab MEKi Chemotherapy Hormone therapy | Breast, pancreatic, neuroendocrine, urothelial, prostate, and renal carcinomas, other solid malignances and ALL |
NVP-BTG226 | Phase I/II | PIK3/mTOR | Breast and other solid malignances | |
PKI-587 (PF-05212384) | Phase I and Phase II | PIK3/mTOR | Endometrial, CRC, and other solid malignances | |
XL765 (SAR25409) | PIK3/mTOR | Erlotinib Temozolomide Hormone therapy | Glioblastomas, breast cancer, and other solid malignances | |
GSK2126458 | Phase I [53] | PIK3/mTOR | Solid malignances |
9.3.2.3 PDK Inhibitors
Despite the potential interest of inhibiting PDK1, a key activator of AKT, little steps have been given in this direction. For instance, OSU-03012 is a derivate of celecoxib, a nonsteroidal anti-inflammatory drug (NSAID) that could potentially target PDK1, but has not reached clinical development [54].
More interesting are the results of another NSAID, aspirin. In vitro aspirin has shown to impair phosphorylation of AKT resulting in decreased downstream signaling leading to cell growth inhibition and induction of apoptosis [55]. However, this action seems to be driven by the inhibition of the cyclooxygenase-2 (COX-2) instead of PDK1. An observational study has confirmed the activity of the drug in preventing recurrences of colon cancer when PI3K mutations are present [56]. This effect seems to be quite specific to aspirin and is not present with other NSAIDs [57].
Unfortunately no clinical trial assessing the combination of aspirin with mTOR inhibitors is ongoing.
9.3.2.4 Akt Inhibitors
Several compounds that aim to inhibit Akt are currently under development. A combination of an Akt plus an mTOR inhibitor not only would inhibit the PI3K pathway at two different steps but additionally would prevent the feedback loop that enhances Akt activity through mTORC2 (Fig. 9.1b). Thus, a synergistic effect could be expected.
Only two trials have explored the possibility of comibining an Akt with an mTOR inhibitor. One of them is a phase I trial adding MK-2206, that blocks Akt2, to ridaforolimus. However, results have not been published yet (NCT01295632). The other is also a phase I trial combining perifosine, a widely studied Akt inhibitor, with temsirolimus in 34 malignant glioma patients [58]. Communicated as poster, thrombocytopenia, cerebral hemorrhage and lung infection were dose-limiting toxicities. Up to two partial responses were observed among 28 evaluable patients, and the schedule was deemed as deserving further studies.
A comprehensive review of Akt inhibitors under clinical development and their studied combinations is provided in Table 9.4.
Table 9.4
Akt inhibitors under clinical development
Communicated clinical trials | Target | Combinations under development | Tumors treated | |
|---|---|---|---|---|
Triciribine (API-2) | Akt 1, 2, 3 | None | Advanced hematologic malignancies, ovarian cancer, cervical cancer, and other solid malignances | |
MK-2206 | Akt2 | Hormonal therapy Chemotherapy Gefitinib Erlotinib Lapatinib Trastuzumab Dalotuzumab Ridaforolimus Selumetinib (MEKi) | AML, CLL, refractory diffuse large B cell lymphoma, hematological malignances, melanoma, ovarian, breast, lung, colorectal, pancreatic, endometrial, head and neck, and liver and kidney cancers and other solid malignances | |
GSK690693 | Phase I | Akt 1, 2, 3 | None | Hematological malignances |
GSK2141795 | Phase I | Akt 1, 2, 3 | Trametinib Dabrafenib GSK1120212 (MEKi) | MM, AML, melanoma, ovarian and endometrial cancer, other solid malignances |
KP372-1 | Preclinical | PDK1, Akt, Flt-3 [72] | None | – |
Perifosine (KRX-0401) | Phase III [91] | Akt | Temsirolimus Imatinib Chemotherapy UCN-01 Bortezomib Lenilamide Sunitinib Sorafenib | MM, AML, CML, lymphomas, myelodysplastic syndromes, Waldenstrom macroglobulinemia, GIST, melanoma, colorectal, prostate, breast, pancreatic, head and neck, kidney, lung, and ovarian cancer, gliomas, soft tissue sarcomas, and other solid malignances |
PBI-05204 (oleandrin) | Phase I [92] | Akt, FGF-2, NF-kappaB, and p70S6K | None | Solid malignances |
RX-0201 | Phase I | Akt expression (anti-sense oligonucleotide) | Chemotherapy | Solid malignances |
9.3.3 Transmembrane Receptors and Ligands
Several transmembrane receptors tyrosine kinases (RTKs) use PI3K as intracellular second messenger. Some of them are “drugable” by compounds that are under clinical development or have even reached daily practice.
The feedback loop that enhances the expression of some of these receptors when mTOR inhibitors are administered has been well documented (Fig. 9.1) leading to the notion that mTOR inhibitors will only work in combination with other drugs [93, 94].
9.3.3.1 ErbB Receptors Family
Three of the four plasma membrane-bound RTKs of the ErbB family have largely been associated to mTOR activity and to resistance to rapalogs (ErbB-1, also known as epidermal growth factor receptor [EGFR], ErbB-2 [or HER2], and ErbB-3 [or HER3]). EGFR and HER2 are known to use both the PI3K/AKT/mTOR and the MAP kinases pathways as second messengers. For this reason, when inhibiting just one pathway, the other will remain active.
Additionally HER2 blockade has been described to enhance HER3 expression probably through a feedback loop where decreased Akt function relieves FOXO that will enhance RTKs translation (Fig. 9.1c) [95].
Altogether, these data point toward a potential synergism between the ErbB receptors inhibitors and mTOR inhibitors. Such combinations are being widely studied not only with rapalogs but with most of the PI3K inhibitors and are one of the most promising lines of investigation for the near future:
HER2
Data from six clinical trials combining everolimus plus a Her2 inhibitor have been already communicated. Five were performed with trastuzumab in breast cancer patients who had progressed on trastuzumab monotherapy and one with lapatinib in a more heterogenous population:
(a)
In 2011 Morrow et al published the results of a pooled analysis of two trials assessing the toxicity and efficacy of the combination of trastuzumab every 3 weeks plus daily everolimus. Forty seven patients were included with an overall response rate (ORR) of 15 %.
(b)
In 2010 Campone et all published the results of a Ib clinical trial combining full doses of weekly paclitaxel (80 mg/m2) plus trastuzumab (2 mg/kg) with escalating doses of everolimus [96]. Standard full dose of everolimus (10 mg daily) was reached and prompted for further development. An impressive ORR was achieved (44 %) with neutropenia and stomatitis as most frequent toxicities.
(c)
Three years later, the same group communicated their experience with such combination in a phase II trial. ORR was 21 % with grade 3–4 neutropenia in 30 % of the cases and stomatitis in 20 % [97]. A phase III trial (named BOLERO-1) has been recently communicated. Though the addition of everolimus to standard trastuzumab and paclitaxel failed to impact progression free survival, a potential role in hormone receptor negative, HER2-positive patients has been suggested [98].
(d)
Another triple combination, vinorelbine plus trastuzumab plus everolimus, was assessed by Jerusalem et al. in a phase Ib trial that included 50 patients. Similarly to the combination with paclitaxel, vinorelbine and trastuzumab were used at standard dose, and everolimus doses were escalated, with the recommended dose being 5 mg daily. ORR was 19 % with apparently lesser neutropenia and stomatitis than with the former schedule, leading to another phase III trial (named BOLERO-3) that compared vinorelbine plus trastuzumab with or without everolimus. Five hundred and sixty-nine patients were included, demonstrating a significant increase in PFS with everolimus (7.00 months [95 % CI 6.74–8.18]) vs placebo (5.78 months [5.49–6.90]) with a hazard ratio 0.78 [95 % CI 0.65–0.95]; p = 0.0067) [99]. Interestingly an exploratory biomarker subanalysis showed that tumors with PTEN deficits or pS6 overexpression (both markers of activation of the PI3K/AKT/mTOR pathway) had better outcome under everolimus.
Finally, a phase I trial has established the dose of lapatinib 1250 mg plus everolimus 5 mg both daily as the recommended dose for phase II studies [100].
Regarding temsirolimus and ridaforolimus, results of trials with trastuzumab have not yet been published.
Epidermal Growth Factor Receptor
There are four approved inhibitors of the EGFR: two small molecules (gefitinib and erlotinib) that inhibit the intracellular tyrosine kinase domain and two monoclonal antibodies (cetuximab and panitumumab) that bind to the extracellular domain.
Regarding everolimus, seven studies have been published about combinations with EGFR inhibitors (three with gefitinib, two with erlotinib, one with cetuximab, and one with panitumumab) and only one regarding temsirolimus (combined with erlotinib) Table 9.5.
Table 9.5
Combinations of mTOR inhibitors with EGFR inhibitors
Reference | Combination CT Phase # of pts | Dose | Comments |
|---|---|---|---|
Kordes et al. [101] | Eve + Cape + Cetuxi I/II 47 | Eve 5 mg/d Cape 600 mg/m2/bid Cetuxi 250 mg/m2/w | Pancreatic cancer DLT: stomatitis, rash, hand-foot syndrome |
Papadimitrakopoulou et al. [102] | Eve + Erlo I 94 | Eve 5 mg/d or 50 mg/w Erlo 150 mg/d | NSCLC DLT: stomatitis, rash, diarrhea |
Vlahovic et al. [103] | Eve + Bev + Pani I 31 | Eve 5 mg tiw Bev 10 mg/kg/Biw Pani 4.8 mg/kg/Biw | Solid tumors DLT: stomatitis, rash, thrombocytopenia |
Bullock et al. [104] | Eve + Erlo + bev I/II 48 | Eve 10 mg/d Erlo 75 mg/d Bev 5 mg/kg/Biw | Solid tumors DLT: stomatitis, rash |
Price et al. [105] | Eve + Gefi II 62 | Eve 5 mg/d Gefi 250 mg/d | NSCLC No relevant activity |
Kreisl et al. [106] | Eve + Gefi II 22 | Eve 5 mg/d Gefi 250 mg/d | GBM No activity |
Milton et al. [107] | Eve + Gefi I 10 | Eve 5 mg/d Gefi 250 mg/d | NSCLC DLT: stomatitis, grade 5 hypotension |
Bauman et al. [108] | Tem + Erlo II 12 | Tem 15 mg/k/w Erlo 150 mg/d | HNSCC Early termination due to toxicity |
Overall, combinations of mTOR plus EGFR inhibitors were poorly tolerated leading to schedules with doses below the standards. Though some phase II trials are still ongoing, the reported results have been disappointing so far, with relevant toxicity and responses rates below 20 % in all cases. Whether this lack of clinical benefit is due to a suboptimal exposure to drugs or just a lack of synergism probably will remain unknown since no major advances are foreseen in this line of investigation.
Thus, expectancies rose by the strong molecular rational of these combinations now rely on other PI3K inhibitors currently under investigation.
9.3.3.2 Insulin-Like Growth Factor Receptor (IGFR)
mTOR inhibitors are known to enhance insulin-like growth factor receptor 1 (IGF-1R) signaling leading to downstream AKT activation [109]. Conversely IGFR inhibition leads to a sensibilization of tumor cells to mTOR inhibitors in vitro, providing a good rational for combining these two types of agents [110].
Everolimus has been combined with figitumumab, an anti-IGFR antibody, in a phase I trial [111]. Full doses of both agents could be administered to all participants and were recommended for a phase II trial. Unfortunately no further studies with figitumumab are foreseen.
Four communications have reported the results of combining temsirolimus plus cixutumumab, another antibody against IGF-1R. In 2011 Naing et al published the original phase I trial that determined the recommended dose for further development [112]. Initial activity in Ewing’s sarcoma and adrenocortical carcinoma was observed; therefore, two extension cohorts focusing in such tumors were prompted [113, 114].
In Table 9.6, results of all five published clinical trials are resumed.
Table 9.6
 The Role of mTOR Inhibitors in Neuroendocrine Tumors
The Evolving Role of Mammalian Target of Rapamycin (mTOR) Inhibitors in Renal Cell Carcinoma
The Role of PI3K/AKT/mTOR Inhibitors in the Treatment of Hematological Malignancies
Potential Future Indication of Rapamycin Analogs for the Treatment of Solid Tumors
The Role of mTOR Inhibitors in Neuroendocrine Tumors
The Evolving Role of Mammalian Target of Rapamycin (mTOR) Inhibitors in Renal Cell Carcinoma
The Role of PI3K/AKT/mTOR Inhibitors in the Treatment of Hematological Malignancies
Potential Future Indication of Rapamycin Analogs for the Treatment of Solid Tumors
 Predictive Biomarkers of Response to mTOR Inhibitors
Predictive Biomarkers of Response to mTOR Inhibitors
 The Clinical Pharmacology and Toxicity Profile of Rapalogs
The Clinical Pharmacology and Toxicity Profile of Rapalogs




Combinations of mTOR inhibitors with IGFR inhibitors
Related posts:
The Role of mTOR Inhibitors in Neuroendocrine Tumors
The Evolving Role of Mammalian Target of Rapamycin (mTOR) Inhibitors in Renal Cell Carcinoma
The Role of PI3K/AKT/mTOR Inhibitors in the Treatment of Hematological Malignancies
Potential Future Indication of Rapamycin Analogs for the Treatment of Solid Tumors
Predictive Biomarkers of Response to mTOR Inhibitors
The Clinical Pharmacology and Toxicity Profile of Rapalogs
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
