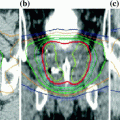
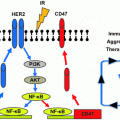
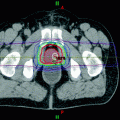
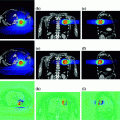
Fig. 1
Ideally, drug combinations should shift the balance between the pro-immunogenic and immunosuppressive effects of radiotherapy to favor its pro-immunogenic effects and or abrogate the immune-suppressive ones. Multiple strategies in each of these two directions are undergoing investigation pre-clinically and clinically
1.1 Immunostimulatory Activity of Radiation Therapy
Radiation therapy has several effects on the immune system.
First, the cell-killing effect of radiation releases a series of signals that are relevant to immune rejection, resulting in cross-priming by DCs. The successful outcome of cross-priming after radiation therapy is limited by the number of intratumoral DCs (Pilones et al. 2014). Radiation is a powerful inducer of immunogenic cell death (ICD), a type of cellular demise that is sensed by the immune system, as it initiates immune rejection. The three hallmarks of ICD include: (1) translocation of calreticulin from the endoplasmic reticulum to the cell surface (Obeid et al. 2007); (2) release of the nuclear protein, high-mobility group box-1 (HMGB1) into the extracellular space and (3) release of adenosine triphosphate (ATP) which activates the inflammasome and causes interleukin (IL)-1β release (Apetoh et al. 2007; Galluzzi et al. 2007; Ghiringhelli et al. 2009; Ma et al. 2010).
Second, radiation therapy leads to DNA release from dying tumor cells. Delivery of tumor DNA to DCs activates the stimulator of interferon genes (STING) pathway and enhances interferon-1 (IFN-1) production by DCs (Deng et al. 2014; Woo et al. 2014). IFN-1 is necessary for the recruitment of DCs to tumors and their subsequent activation (Diamond et al. 2011; Fuertes et al. 2011). Activated DCs migrate to draining lymph nodes where they cross-present tumor-derived antigens to T-cells, resulting in anti-tumor T cell responses.
Moreover, radiation therapy promotes the release of chemokines that attract T-cells to tumors, enhancing trafficking. Studies revealed that radiation therapy induces expression and release of the chemokines CXCL10 and CXCL16 (Lugade et al. 2008; Matsumura et al. 2008). CXCL16 is a chemokine that binds to CXCR6 on Th1 and activated CD8+ effector T cells, and plays an important role in T-cell recruitment to sites of inflammation. Using 4T1, a poorly immunogenic mammary cancer murine cell line, Matsumura et al. (2008) demonstrated that irradiation of cells in vitro caused an over fourfold increase in CXCL16 mRNA. This effect peaked at 48 h after irradiation. In addition, when injected in a syngeneic immune-competent mouse, irradiation (12 Gy × 2) of 4T1 tumors induced CXCL16 in blood vessels and on the majority of tumor cells at immunohistochemistry, 48 h after tumor irradiation. In comparison, for un-irradiated tumors, immunohistochemistry for CXCL16 yielded only weak baseline CXCL16 immuno-reactivity in selected vessels and faint staining in tumor cells. These results provide evidence that irradiation can induce the secretion of key pro-inflammatory chemotactic factors that recruit antitumor effector T cells to the irradiated field.
Radiation therapy also increases the expression of major histocompatibility complex (MHC) class I proteins, necessary for antigen recognition by cytotoxic CD8+ T cells (CTLs) (Reits et al. 2006). MHC class I displays fragments of non-self antigens to cytotoxic T cells through the cytosolic/endogenous pathway. This effect of radiation is particularly important since tumors commonly escape recognition by the immune system via down-regulation or loss of the MHC-I molecules. In the GL261 preclinical mouse model of intracranial glioma, radiation therapy combined with granulocyte macrophage colony stimulating factor (GM-CSF) enhanced tumor infiltration by T cells and reversed MHC-I down-regulation of invasive glioma cells (Newcomb et al. 2006). In this model peripheral vaccination with GL261 tumor cells and GM-CSF (without radiation) resulted only in minimally improved survival of the treated mice compared to control animals. Conversely by recovering this basic mechanism of cross-presentation, radiation led to long-term survival in 80% of vaccinated mice, who also rejected challenge with the same tumors.
Similarly, radiation therapy increases the expression of Fas/CD95 (Chakraborty et al. 2003) and adhesion molecules which also participate in the mechanism of tumor cells recognition and elimination by CTLs. Radiation therapy also induces surface expression of NK group 2, member D (NKG2D) ligands such as UL-16 binding proteins, Rae-1 and MICA/B, which mediate tumor cell killing by both CTLs and natural killer cells (Kim et al. 2006). Tumor killing via NK cells is especially important when tumors have lost key components of the MHC-I complex rendering them unrecognizable by CTLs. Apart from direct cell killing, NK cells also enhance radiation sensitivity. Incubation with human NK cells prior to irradiation led to greater growth inhibition, radiation cytotoxicity and apoptosis among tumor cells from a variety of malignancies (Yang et al. 2013). Experiments using nasopharyngeal cancer cells revealed that incubation with NK cells led to an increase in the level of cytosolic Granzyme B, the molecule that initiates the proteolytic caspase cascade to cause cell death. Without radiation therapy, X-linked inhibitor of apoptosis protein (XIAP) inhibits apoptosis via the intrinsic mitochondrial apoptosis pathway by binding caspase-3. However, in the presence of radiation therapy, Smac is released from mitochondria and forms a complex with XIAP to enable Granzyme B-induced apoptosis.
Another radiation-induced mechanism that improves T-cell trafficking to the tumor involves reprogramming of the established immunosuppressive microenvironment by tumor-associated macrophages (TAMs). At baseline, most TAMs express the M2 phenotype (Stout et al. 2005), which promotes angiogenesis, tumor growth and metastasis while impairing T-cell function. In a mouse pancreatic cancer model, a single 2 Gy dose of radiation therapy caused significant expression of inducible nitric oxide synthetase (iNOS) in TAMs, eliciting what has been termed the M1 response (Klug et al. 2013). iNOS metabolizes L-arginine to produce L-citrulline and nitric oxide which promote immune and inflammatory reactions (Bansal and Ochoa 2003; Boucher et al. 1999). As a result, tumor macrophages normalized the tumor vasculature, recovering tumor perfusion and overcoming the barrier to T-cell infiltration of cancer-induced, abnormal vasculature.
Additional immunostimulatory effects of radiation therapy include upregulation of ICAM-1 (Ruocco et al. 2012), vascular cellular adhesion molecule 1 (VCAM-1) on tumor endothelium, which facilitates tumor infiltration by T, cells that produce interferon γ (IFN-γ) and tumor necrosis factor α (TNF-α).
1.2 Immunosuppressive Effects of Radiation Therapy
Although radiation therapy can function as an effective adjuvant for immunotherapy, it also has many identified immunosuppressive effects.
In contrast to the beneficial effects of low-dose radiation mentioned above, whereby a single low dose of radiation therapy induces tumor infiltration by TAMs that express iNOS, radiation also induces the expression of immunosuppressive enzymes such as arginase-1 and cycloxygenase-2 (COX-2) in TAMs (Tsai et al. 2007). Arginase converts L-arginine to polyamine precursors that can function as tumor growth factors (Chang et al. 2001). Moreover, depletion of L-arginine causes decreased expression of the T-cell receptor (TCR) signaling zeta chain (CD3zeta), impairs T cell proliferation and decreases cytokine output (Rodriguez et al. 2003). Similarly, COX-2 expression is associated with increased proliferation, invasion and angiogenesis (Fujita et al. 2002; Attiga et al. 2000; Wang et al. 2005). In murine prostate cancer cells, a single dose of 25 Gy led to a transient 1.4 and 2.3-fold increase in Arg-I and COX-2 mRNA, respectively, at 8 h. Beginning day 3, the Arg-I and COX-2 levels steadily rose to 1.5-fold and 5.6-fold, respectively, at 3 weeks. Increased iNOS expression began 3 days after irradiation and peaked at over 6-fold at 3 weeks. After fractionated radiation therapy (60 Gy in 15 fractions), COX-2 and Arg-I mRNA expression increased by the fifth fraction and was at least three-fold higher by the end of treatment. iNOS expression was not increased by the 10th fraction and only rose minimally by the final fraction (1.3 fold). Of note, low dose iNOS can promote angiogenesis in tumors while high doses have tumoricidal effects (Jenkins et al. 1995). These data suggest that the commonly used fractionated radiation therapy regimens may induce immune suppressive responses in tumors.
Importantly, single ablative doses of radiation (>10 Gy in one fraction) results in extensive endothelial cell death that may reduce vascular flow and impair effector T-cell trafficking to the tumor (Park et al. 2012). In addition, single high dose radiation therapy has been shown to promote a hypoxia-driven immunosuppressive environment in mouse melanoma models (Park et al. 2012; Hasmim et al. 2013).
Fractionated radiotherapy in classical therapeutic doses has been shown to increase additional mediators of immune suppression. First, it increases infiltration by myeloid-derived suppressor cells (MDSCs). This effect is mediated by colony stimulating factor 1 (CSF1), a chemokine that recruits MDSCs. In human and mouse prostate cancers, DNA damage caused by irradiation triggered ABL1 protein translocation to the nucleus and binding to the CSF1 promoter. These changes resulted in increased CSF1 gene transcription, which led to an increase in circulating MDSCs (Xu et al. 2013). Regulatory T cells are also increased after radiation therapy (Bos et al. 2013). These cells play a role in maintaining tolerance and suppressing antitumor immunity. Depletion of regulatory T-cells enhances the growth inhibitory effects of radiotherapy in murine carcinoma models (Bos et al. 2013).
Radiation is a powerful activator of transforming growth factor (TGF) β, that also contributes to immunosuppression. Reactive oxygen species, created by radiation therapy, cause TGFβ to dissociate from latency-associated peptide (LAP) (Barcellos-Hoff et al. 1994). In its active form, TGFβ inhibits stimulation of DCs and reduces priming of CD8+ T cells. In mouse breast carcinoma models, these inhibitory effects were overcome using antibodies that neutralized TGFβ (Vanpouille-Box et al. 2015). In mice bearing 4T1 breast cancers, intraperitoneal injection of 1D11, a TGF β neutralizing antibody, prior to irradiation led to a significant increase in the percentage of DCs, CD4+ and CD8+ T cells infiltrating the irradiated tumors. Moreover, in mice bearing two tumors, the combination of RT and 1D11, led to increased T cells infiltration of non-irradiated tumors suggesting that TGF β blockade may enable abscopal responses.
Finally, recent evidence implicates galectin-1, a carbohydrate-binding protein, as a barrier to the immune-mediated response to radiotherapy. In a syngeneic mouse model of non-small cell lung cancer, radiation induced galectin-1 secretion, leading to lymphopenia, due to a decrease in circulating CD3+ and CD8+ T cells through T-cell apoptosis (Kuo et al. 2014). These immune-suppressive effects were prevented with the use of thiodigalactosidase, a Gal-1 competitive inhibitor, or an anti-Gal-1 specific antibody.
Strategies to abrogate the immunosuppressive responses to radiation therapy may be necessary in order to best harness its potential for contributing to antitumor immunity.
2 Pre Clinical and Clinical Combinations of Radiotherapy and Immune Agents
2.1 Toll-like Receptor (TLR) Agonists
At the time tumors are discovered and the patient is diagnosed with cancer, multiple immunosuppressive mechanisms are in place that maintain their growth, making the tumor microenvironment immune-privileged (Joyce and Fearon 2015). For instance, macrophages, MDSCs and DCs, capable of suppressing T-cell activation, are often present in tumors.
Toll-like receptors (TLRs) activate innate immunity and initiate adaptive immune responses when stimulated by pathogen-derived and/or endogenous ligands (Adams 2009). Administration of synthetic TLR agonists has been shown to overcome some of the existing immunosuppressive barriers in established tumors by enhancing DC stimulation. Unmethylated C-G motifs (CpG) are single strand oligodeoxynucleotides that contain cytosines and guanines and they function as TLR9 agonists. They exert their effects on plasmacytoid DCs to induce IFN-α production, antigen presentation and upregulation of costimulatory molecules (Vollmer et al. 2004). They also stimulate cytokine production from Th1 cells (Wooldridge and Weiner 2003). In preclinical experiments, incubation of CpG with lymphoma cells led to the expression of costimulatory molecules, CD80 and CD86 as well as inhibition of proliferation (Li et al. 2007). The combination of intraperitoneal chemotherapy and intratumoral CpG injection in mice bearing B-cell lymphomas activated a CD8-dependent T cell immune response against local and systemic tumors, delayed tumor recurrence and prolonged survival of the mice. These results prompted a Phase I/II study of 15 patients with relapsed low-grade B-cell lymphoma (Brody et al. 2010). The study was designed to harness the anti-tumor vaccination properties of radiotherapy and intratumoral TLR9 in order to induce abscopal effects. A CpG-enriched TLR9 agonist was injected into the tumor site immediately before the first radiation therapy treatment, after the second treatment then weekly for 8 consecutive weeks. Radiation therapy consisted of 4 Gy administered consecutively daily in 2 Gy fractions. After a median follow-up of 33.7 months, there was one complete response, three partial responses and eight patients with stable disease for an overall response rate of 27%. Results were durable; the complete response lasted for 61 weeks while the 3 partial responses were maintained for 29, 64 and 111 weeks. Similarly, several patients had stable disease for up to 131 weeks. Like other immunotherapeutic treatments, clinical response peaked after ≥24 weeks. These remarkable results were obtained in a cohort of patients with a median of 3 prior failed therapies (range 1–6). Of note, the patient with a partial response lasting 64 weeks was re-treated with a higher dose of the TLR9 agonist, and then achieved a second PR within 12 weeks from re-treatment, which was faster than the initial PR that had occurred after 43 weeks. The development of flu-like symptoms during therapy and fewer prior therapies correlated with a greater magnitude of clinical response.
A series of immunological studies were conducted in this trial to elucidate the relationship with the clinical responses. These tests revealed a negative relationship between Treg induction and clinical response. Pre-treatment peripheral blood lymphocytes (PBLs) were cultured with autologous, irradiated, CpG-activated tumor B cells. At baseline, Treg levels among pre-treatment PBLs were low, with an average of 7.3%. After culture with autologous tumor cells, Treg proportion increased to an average 19.7% and was greater with CpG-activated tumor cells than with untreated tumor cells. The range of Treg increase varied, with 5 patients eliciting at least 4-fold increase and 9 patients eliciting ≤2-fold increase. Non-Treg inducers tended to have better clinical responses and significantly longer progression-free survival than Treg inducers. Interestingly, the baseline proportion of Tregs in patients’ tumors and peripheral blood did not correlate with clinical outcomes, suggesting that it is the plasticity of Treg response after TLR agonists that matters. Additional experiments to modify Tregs include intratumoral injections of anti-CTLA-4 antibodies (Marabelle et al. 2013).
PBLs were also co-cultured with CpG-activated autologous tumor cells then assessed for activation markers such as CD-137, IL-2, interferon-γ and tumor necrosis factor. In some patients, disease regression correlated with improving immune response. However, these results were not statistically significant nor did all clinically responsive patients demonstrate tumor-reactive CD8+ T-cells.
The clinical availability of Imiquimod, a TLR agonist that specifically activates TLR7, which is expressed by both plasmacytoid DCs and CD11c+ myeloid-derived DCs, has made it an ideal candidate for clinical investigation (Stanley 2002). For instance, the combination of radiation therapy and TLR agonists is also being explored in breast cancer. In a preclinical syngeneic model using poorly immunogenic TSA mouse breast carcinoma cells, mammary adenocarcinoma were implanted subcutaneously, under the mouse skin, to mimic a chest wall recurrence of breast cancer (Dewan et al. 2012). Low-dose cyclophosphamide was delivered intra-peritoneally prior to the topical application of 5% imiquimod versus placebo cream to the skin overlying tumors, three times a week. A distinct subset of mice were also treated with cyclophosphamide which also reduces the proportion of circulating Tregs, to test the additional effect of this immune therapy. Radiation therapy was initiated 12 days after tumor injection and delivered in three consecutive daily fractions of 8 Gy. Either radiation therapy or imiquimod as single modalities resulted in some delay of tumor growth. However, radiation therapy in combination with imiquimod led to the regression of the majority of the tumors between days 25 and 30 as well as improved survival of the experimental animals, demonstrating synergy of the combination. Other experiments demonstrated that application of imiquimod and radiotherapy to the primary tumor led to tumor growth inhibition at a secondary un-irradiated site that had also been inoculated with TSA cells. Imiquimod/irradiated tumors demonstrated increased expression of two MHC class I alleles, intercellular adhesion molecule 1 (ICAM1) and infiltration by CD11c+ DCs, CD4+ and CD8+ T cells. Depletion of CD8+ T-cells suppressed these effects. Both CD8+ T-cells and CD8+ presenting DCs were key to the success of the combination that promoted both the priming and effector phases of anti-tumor T-cell responses. Importantly, when responding mice treated with low dose cyclophosphamide (a drug that reduces the number of regulatory T cells), radiation therapy and imiquimod were re-challenged with TSA cells after 90 days, they failed to develop tumors, showing long-term immunologic memory.
Based on these findings, an ongoing Phase I/II study of imiquimod, cyclophosphamide and radiation therapy for patients with breast cancer dermal or chest wall metastases is being conducted (ClinicalTrials.gov identifier: NCT01421017).
A previous Phase II trial in ten patients of single modality imiquimod for chest wall recurrences of breast cancer demonstrated a 20% ORR (Adams et al. 2012; Demaria et al. 2013). The current Phase I/II trial attempts to improve local and systemic anti-tumor immune response via the synergistic combination of imiquimod and radiation therapy. Radiation therapy is delivered to 1 area of skin metastases in 5 fractions of 6 Gy on Days 1, 3, 5, 8, and 10. As in the previous Phase II trial, imiquimod cream is applied topically 5 nights per week for 8 weeks, beginning on Day 1. During the Phase I portion, 6 patients completed treatment without any dose limiting toxicities. Phase II is currently underway with a target accrual of 25 additional patients. The primary endpoint is the response rate in untreated distant metastases, which will be assessed by immune-related response criteria. The local tumor responses, safety of the combination, immune-mediated rejection signatures and peripheral lymphocytes for antigen-specific T and B cell responses are also analyzed.
Imiquimod in combination with radiation therapy is also being used in a pilot study in diffuse intrinsic pontine glioma, a pediatric brain tumor with a poor prognosis (ClinicalTrials.gov identifier: NCT01400672). Patients will first receive 55.8 Gy over the course of 6–7 weeks directed to the intracranial tumor. Four weeks later, they will receive the first of 4 intradermal vaccines produced using the brain tumor initiating cell line GBM-6 as the antigen source. Vaccine will be injected at two separate sites every two weeks for 4 doses then every 4 weeks for up to 1 year. Imiquimod will be applied to the two vaccination sites 24 h after each injection. At the time of the 1st and 3rd vaccinations, 180 cGy fractions (for a total of 59.4 Gy) will be administered to the intracranial tumor with the intent to upregulate NKG2D ligands and enhance tumor killing by CTLs and NK cells.
2.2 Cancer Vaccines
In a Phase II clinical trial involving 30 prostate cancer patients with localized disease, the participants were randomized to receive standard definitive radiotherapy alone or in combination with a poxviral vaccine encoding prostate-specific antigen (PSA) (Gulley et al. 2005). Patients on the combination arm received recombinant vaccinia (rV) PSA plus rV containing the T-cell costimulatory molecule B7.1 (rV-B7.1) followed by monthly booster vaccines with recombinant fowlpox PSA. The vaccines were given with local GM-CSF and low-dose systemic interleukin-2. Standard external beam radiation therapy was given between the fourth and the sixth vaccinations. Overall, treatment with the combination was well tolerated. Of nineteen patients enrolled on the combination arm, seventeen patients completed all eight vaccinations. An increase in PSA-specific T cells of at least 3-fold was noted in 13 of 17 patients. No T-cell increases were detected in patients on the radiotherapy-only arm. There was also evidence of de novo generation of T cells to well-described prostate-associated antigens that were not part of the vaccine. This observation suggests that radiation therapy enabled the mechanism of antigenic spread, enabling vaccine-elicited T-cells to better access the tumor, and induce T-cell mediated killing, with release of additional antigens and priming of new T-cell reactivity. These cascading effects support the role of combining radiotherapy with tumor- directed vaccines.
2.3 GM-CSF
Similar to the experiments already described with Flt3-L used to stimulate DC production during radiation (Demaria et al. 2004), GM-CSF can also potentiate cross-presentation of antigens released from the irradiated tumor. T cells, macrophages, endothelial cells and fibroblasts secrete GM-CSF in response to immune stimuli. At low concentrations, GM-CSF stimulates macrophage proliferation while moderate concentrations elicit dendritic cell proliferation and maturation (Burgess and Metcalf 1980).
To translate our preclinical experience with Flt3-L (2) to the clinic, we designed a proof-of-principle trial that substituted Flt3-L with GM-CSF, which was available for clinical use. Abscopal responses were detected in 26.8% of patients with metastatic cancer who received radiotherapy (35 Gy in 10 fractions) and concurrent GM-CSF injected subcutaneously daily for two weeks, beginning with the second week of radiotherapy (Golden et al. 2015). Median overall survival was significantly better in abscopal responders than in patients without abscopal responses (20.98 months vs. 8.33 months). Of note, abscopal responders presented with lower baseline median neutrophil to lymphocyte ratio than non-responders (2.29 vs. 4.24). This finding is consistent with previous reports that a neutrophil to lymphocyte ratio greater than 4 is a poor prognostic marker (2014). Overall, treatment was well tolerated. The most common side effects were Grade 1 fatigue (35 patients), Grade 1 dermatitis (13 patients) and Grade 1 nausea/vomiting (19 patients). Importantly, this trial demonstrates that despite advanced metastatic disease and extensive pretreatment, patients can derive benefit from localized radiation and immunotherapy.
2.4 TGF-β Antagonist
As previously described, reactive oxygen species created by radiation therapy cause TGFβ to dissociate from its latency-associated peptide (LAP) (Barcellos-Hoff et al. 1994). TGFβ induction has multiple concurrent effects: it promotes DNA damage response and modulates radiosensitivity (Bouquet et al. 2011), and inhibits the antigen-presenting function of DCs (Wrzesinski et al. 2007). In preclinical models of metastatic breast cancer, radiation therapy combined with TGFβ neutralizing antibodies induced T-cell mediated rejection of the irradiated tumor as well as the un-irradiated metastases (Vanpouille-Box et al. 2015). Of note, neither TGFβ blockade nor radiation therapy alone had an effect on lung metastases. Conversely, radiation combined with TGFβ blockade led to complete regression of 81% of the primary irradiated tumors as well as significant growth inhibition of contralateral non-irradiated tumors and lung metastases. Microarray analysis of primary tumors treated with radiation therapy and TGFβ blockade revealed that the top 20 upregulated pathways were immune-related and the top 3 gene networks were involved in recruitment of CTLs and the activation of immune effector function genes and IFNγ pathways. Finally, use of PD-1 blocking antibody in addition to radiation therapy and TGFβ blockade improved the rate of complete regression (75%) and enhanced survival rates, compared to radiation therapy with TGFβ blockade (44%) or with anti-PD-1 (25%), respectively.
These findings suggest that TGFβ blockade enables CD8+ T cell priming, which improves both local and distant tumor control but, optimal preclinical results could only be achieved by overcoming adaptive immune resistance mediated by PD-L1 expression, supporting a therapeutic strategy that targets multiple immune pathways in combination with radiation therapy. We recently completed a Phase 2 trial investigating the combination of radiation therapy and fresolimumab, a human monoclonal TGFβ antibody, is underway in metastatic breast cancer patients. The primary endpoint is the abscopal response rate at 15 weeks (ClinicalTrials.gov identifier: NCT01401062). After completion of this trial, we plan to test the addition of anti-PD1 to the combination.
2.5 CTLA-4 Blockade
Cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) or CD152 is a costimulatory molecule that is expressed on T-cells. It functions as an immune checkpoint, to down-regulate an immune response. Anti-CTLA4 predominantly inhibits T-regulatory cells (Treg cells), thereby increasing the CD8 T-cell to Treg (CD8/Treg) ratio. Preclinical models of breast and colon carcinoma have demonstrated synergy between anti-CTLA-4 antibody and radiation therapy (Demaria et al. 2005; Dewan et al. 2009). In 4T1 breast carcinoma murine models, anti-CTLA-4 antibody alone did not affect primary tumor growth or mouse survival; radiation therapy only delayed tumor growth without influencing survival (Demaria et al. 2005). However, the combination of radiation therapy and anti-CTLA-4 antibody led to local tumor growth inhibition as well as inhibition of lug metastases and improved survival. Experiments by Ruocco et al. (2012) revealed one of the mechanisms underlying these observations. Anti-CTLA-4 antibody alone enhanced T cell motility and reduced contact time with tumor cells. However, combining anti-CTLA-4 antibody with radiation therapy promoted MHC-I and NKG2D dependent CD8+ T cell arrest in contact with tumor cells and inhibited cell growth (Ruocco et al. 2012). Expression of the NKG2D ligand retinoic acid early inducible-1 (RAE-1) was increased in irradiated 4T1 cells, enabling a more effective immunological synapse.
Clinical translation of the synergy of radiation and anti-CTLA-4 blockade has been reported with cases of abscopal responses in patients with melanoma and non-small cell cancer who received both radiation and ipilimumab, a monoclonal antibody against CTLA-4 (Golden et al. 2013; Grimaldi et al. 2014; Hiniker et al. 2012; Postow et al. 2012). Ipilimumab and irradiation of a liver metastasis elicited a dramatic response within the radiotherapy field and at distant metastases in a 64-year-old man with metastatic lung adenocarcinoma (Golden et al. 2013). Despite multiple lines of chemotherapy and radiation to the chest, the patient was rapidly progressing with multiple metastases in the lung, liver and bones. Radiation therapy to a metabolically active hepatic metastasis was treated with 30 Gy in 5 fractions, with Ipilimumab administered the day after the first radiation fraction then every three weeks for a total of 4 infusions. Four months after treatment, the irradiated lesion and all other metastases had dramatically decreased, and eventually resolved. The patient has remained disease free 3 years later, without any other treatment.
Recently, a phase I dose escalation trial in patients with metastatic melanoma demonstrated partial responses in 18% of study participants and stable disease in another 18% (Twyman-Saint Victor et al. 2015). Patients with lung or bone metastases received 8 Gy × 2 or 8 Gy × 3 to an index lesion while those with liver or subcutaneous metastases received 6 Gy × 2 or 6 Gy × 3. Three to five days after radiation therapy, all patients received ipilimumab every three weeks for four doses. Preclinical models revealed that resistance to this treatment combination was due to T-cell exhaustion and upregulation of PD-L1 on melanoma cells. Among patients on this trial, high PD-L1 expression in the melanoma cells prohibited response to the treatment regimen and was associated with rapid disease progression and persistent T-cell exhaustion. While the findings could also demonstrate PD-L1 expression as a sign of T cell activation, it is possible that anti-CTLA4 and anti-PD-L1/PD-1 therapies may need to be combined with radiation in order to improve systemic response.
At present, there are multiple trials underway that explore the combination of radiation therapy and ipilimumab. One example is a Phase 2 trial in metastatic non-small cell lung cancer patients. Patients receive ipilimumab within 24 h of starting radiation therapy (6 Gy × 5 or 9.5 Gy × 3) to an index lesion. Ipilimumab is given every three weeks for a total of four doses. The primary endpoint is the abscopal response rate (ClinicalTrials.gov identifier: NCT02221739).
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


