6 Radiotherapy and Chemotherapy
Exploitable Strategies in Combining Chemotherapy With Radiation
The primary objective of combining chemotherapy with radiation is to achieve an improved therapeutic result, which can be evaluated as a function of enhanced tumor response or reduced normal tissue toxicity. In the late 1970s, Steel and Peckham developed a conceptual framework for analyzing drug-radiation interactions.1 In this seminal work, four mechanisms were described in which combined modality therapy could improve therapeutic outcome: spatial cooperation, toxicity independence, protection of normal tissue, and enhancement of tumor response. For more than 20 years, these mechanisms provided the backbone for evaluating chemoradiation combinations clinically. Based on lessons learned from these clinical investigations, coupled with the rapid emergence of molecularly targeted agents, Bentzen and colleagues proposed an updated conceptual framework to evaluate drug-radiation combinations, which are summarized in the following text and in Fig. 6-1.2
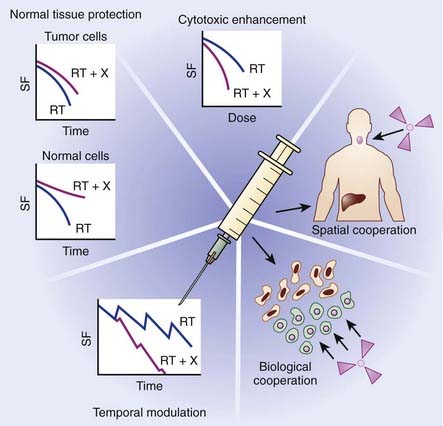
Spatial cooperation refers to combining a drug that is efficacious against systemic disease with radiation, which is effective against locoregional disease. Because a full dose of radiotherapy and chemotherapy is required, and spatial cooperation does not require an interaction at the cellular level, these modalities are typically administered sequentially in an effort to reduce toxicity. Examples of spatial cooperation include adjuvant chemotherapy and radiation therapy in breast cancer and the combination of hormonal therapy and radiation in patients with high-risk prostate cancer. Although this approach is advantageous in terms of toxicity, the issue of overall treatment must also be considered in the design of these schedules. By integrating a chemotherapeutic agent either before or after definitive radiation therapy, the overall treatment time would consequently increase, potentially contributing toward accelerated cellular repopulation and diminished response.3,4 For example, a recent overview of randomized controlled trials of combined chemoradiation for small cell lung cancer concluded that shorter time from the start of any cytotoxic chemotherapy to the end of radiotherapy was significantly correlated with improved treatment outcome.5 Similar findings have been identified for head and neck cancer.
Cytotoxic enhancement refers to the capacity of chemotherapy to interact with radiation and produce a greater effect on the local tumor than would be expected from simple additivity of cell killing. A drug with additive cell killing properties might be seen as a borderline case of cytotoxic enhancement, but unless it possesses active biologic targeting, it will not provide an advantage over a simple radiation dose escalation. With the rapid development of molecular oncology, which has identified numerous molecularly targeted agents that target tumor-specific biologic targets, the identification of tumor-specific agents is being actively investigated. Interestingly, preclinical models suggest that many of these agents also appear to enhance radiation response. Numerous trials are currently underway in multiple tumor types to determine the capacity of these agents to serve as radiation sensitizers. The mechanism that underlies this favorable interaction likely involves induction or repair of radiation-induced cellular deoxyribonucleic acid (DNA) damage. For example, incorporation of halogenated pyrimidines such as 5-fluorouracil into DNA seems to enhance the initial induction of DNA damage by radiation.6 The diverse and complex biologic processes that may be targeted by chemotherapy occurring during the interval between fractionated radiotherapy, including tumor-cell repopulation, reoxygenation, and cellular redistribution, have been collectively termed temporal modulation. For example, combining epidermal growth factor receptor (EGFR) blockade with fractionated radiation may reduce cellular proliferation during therapy, thereby attenuating radiation-induced accelerated cellular proliferation.7
Both toxicity independence and normal tissue protection were initially described by Steel and Peckham as exploitable strategies of chemoradiation; however, lessons learned from clinical investigations over the years suggest these mechanisms may no longer have direct clinical relevance. Toxicity independence refers to the concept of combining a drug that caused systemic toxicity with radiation, in which toxicity is expressed locally. It was suggested that this combination could allow for treatment intensification without unacceptable toxicity, even if the anticancer effects of the two modalities were simply additive. Although this approach was eagerly pursued, a number of studies have shown that effective radiochemotherapy combinations did, in fact, increase radiation-related side effects.8–10 Normal tissue protection was initially based on the potential of some agents, such as cyclophosphamide and methotrexate, given before radiation, to reduce the effect of a subsequent irradiation in some normal tissues, including bone marrow and intestinal epithelium. This effect of dosing chemotherapy before radiotherapy is now understood to be a result of induced cellular repopulation after the first cytotoxic insult. Unfortunately, attempts to exploit this mechanism clinically have been unsuccessful. However, normal tissue protection is continuing to be actively investigated using agents specifically designed to provide cytoprotection or modulate the cytotoxic response of normal tissue. A number of strategies in preclinical or early clinical testing fall under this category, such as the stimulation of stem-cell proliferation in early responding normal tissues,11,12 for example the use of keratinocyte growth factors to ameliorate mucositis,13 or cytoprotection using the thiol-based free radical scavenger amifostine.14,15
Advances in molecular oncology have fostered the development of the mechanism termed biologic cooperation, which has been proposed by Bentzen and colleagues to refer to strategies that target distinct cell populations, or employ different mechanisms for cell killing or delaying tumor regrowth.2 An example could be a drug that targets hypoxic tumor cells, thereby complementing the effect of radiation, which has greater response in well-oxygenated cells. This approach has been applied with such agents as misonidazole, nimorazole, tirapazamine, and mitomycin C, which target hypoxic tumor cells either as radiation sensitizers or bioreductive cytotoxins.16 More recently, clinical interest has focused on agents that affect hypoxia indirectly by targeting the tumor microenvironment. For example, vascular-targeted antiangiogenic agents such as combretastatin cause a shutdown of tumor vasculature, leading to tumor cell death via hemorrhagic necrosis. Agents such as combretastatin are mainly effective in the central regions of the tumor where hypoxic radioresistance may be a therapeutic challenge. Biologic cooperation arises because radiation is particularly effective against the well-oxygenated cells in the periphery of the tumor where vascular shut-down is less effective. Biologic cooperation can even be applied when combining angiogenic agents with radiation. For example, Jain recently presented the case of antiangiogenic agents inducing a normalization of tumor blood vessels, which in turn reduced tumor hypoxia, creating potential biologic cooperation between this class of drugs and radiation.17
Methods for Assessing and Defining Drug–Radiation Interactions
Preclinical studies are typically initiated in vitro, using cell-culture systems. With respect to assays that may be used, the clonogenic assay, which detects all forms of radiation-induced cell death, is considered the “gold standard” for radiosensitivity analysis.18 The underlying biologic end-point evaluated in this assay involves reproductive integrity. Following irradiation, a cell may be physically present and apparently intact, and may even progress through one to two cycles of mitosis; however, if it has lost the capacity to divide indefinitely and produce a large number of progeny, it loses its reproductive integrity. In the context of radiation treatment, losing reproductive integrity prevents continued growth and metastases; therefore, from a practical perspective, the tumor is eradicated. A surviving cell, however, retains its reproductive integrity and is able to proliferate indefinitely to produce a large clone or colony, referred to as clonogenic. In the clonogenic assay, cell survival is determined as a function of radiation dose, with the surviving fraction of cells (colonies) plotted on a logarithmic scale and the dose of radiation plotted linearly. To assess the effect of a drug on cell radiosensitivity, the combined drug–radiation curve is commonly plotted after the cytotoxicity produced by the drug alone is excluded, referred to as normalization. The radiation cell-survival curve is not changed if the drug does not influence cell radiosensitivity, regardless of whether the drug is cytotoxic on it own. In this case, the cytotoxicity of the drug contributes only to the overall cell killing by the combined treatment of both agents, referred to as an additive effect. Investigational agents may interact with radiation by altering cell radiosensitivity such that the combination results in a supra-additive or subadditive effect, depending on whether the cell killing is greater or smaller than the sum of cell killings produced by individual agents. This type of interaction often changes the shape of the cell-survival curve. For instance, a modification of the shoulder region indicates an interaction affecting the repair of radiation-induced DNA damage. An example of a clonogenic survival experiment demonstrating the capacity of an investigational agent to enhance radiation response is depicted in Fig. 6-2A.
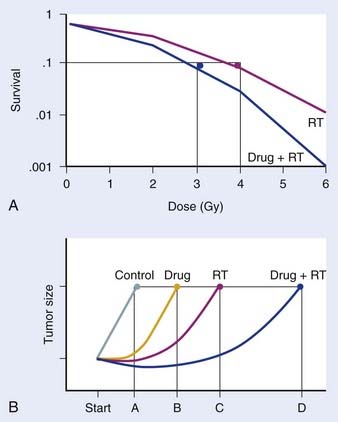
Favorable findings in vitro are often followed by in vivo exploration of drug–radiation interactions. A commonly applied technique involves a mouse xenograft model, in which human tumor cells are grown in culture and inoculated in the flank region of immunodeficient mice. The efficacy of treatment is then determined by the extent of tumor growth delay19 or the rate of tumor cure.20 When the treatment endpoint is delay in tumor growth in drug and radiation combinations, typically four treatment arms are required (control, radiation alone, drug alone, and drug and radiation combined), and tumors are followed until they meet predetermined size criteria. As in the clonogenic assay, growth delays are normalized by subtracting delays of independent treatments (radiation alone and drug alone) from the difference between the drug–radiation combination and the untreated control arm. An example of a growth delay experiment is depicted in Figure 6-2B. The 50% tumor control dose (TCD50; i.e., the dose of radiation that achieves 50% tumor control) assay is used when the treatment endpoint is rate of tumor cure. In this experiment, tumor cure is measured as a function of radiation dose, and the dose that is required to cure or control 50% of the tumors are compared between mice treated with radiation alone and those treated with combined treatment. If combined treatment displaces the curve to the left (i.e., a lower TCD50), then addition of the drug improves tumor curability21; however, this assay does not discriminate between independent drug activity and a direct influence on radiation response. The TCD50 assay has been suggested to be a more relevant model to assess the curative potential of radiation when combined with an investigational agent, as its primary endpoint involves clonogenic cell death.22
Although these in vivo methodologies certainly have their limitations, their potential advantage over traditional in vitro studies is that they also provide insight into the exploitable strategy of biologic cooperation, because both fractionation and tumor microenvironment are evaluated. In an effort to better replicate tumor environment for a specific tumor type, this assay may be of further value if performed orthotopically (e.g., brain tumor grown intracranially, breast tumor grown in mammary fat pads, etc.).23–25 It has been historically suggested that this model may be applied to address normal tissue toxicity; however, its overall clinical application has been limited thus far. Therefore, it is of great importance to identify novel model systems that may be used in this context, because this would provide insight into an improved therapeutic ratio, rather than solely cytotoxic enhancement, which would be the necessary endpoint to further clinical gains.
Initial Radiation Damage
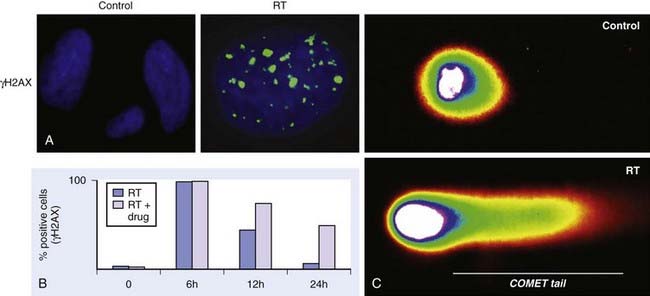
The primary methodology employed to assess radiation damage preclinically is the neutral comet or single-cell gel electrophoresis assay, which is a rapid and sensitive method for detecting DNA damage at the level of individual cells.26 It is based on the ability of negatively charged loops or fragments of DNA to be drawn through an agarose gel in response to an electric field. The extent of DNA migration is directly influenced by the amount of DNA damage present in the cell. In this assay, a suspension of cells are mixed with agarose and spread onto a microscope glass slide. DNA unwinding and electrophoresis is carried out at a specific pH. Unwinding of the DNA and electrophoresis at neutral pH (termed neutral comet) predominantly facilitates the detection of DSBs and crosslinks, whereas, when performed at pH levels greater than 12 (termed alkaline comet), the test facilitates the detection of SSBs and DSBs, incomplete excision repair sites, and crosslinks. When subjected to an electric field, the DNA migrates out of the cell in the direction of the anode, appearing like a “comet” with a distinct head composed of intact DNA, and a tail consisting of damaged or broken pieces of DNA. The size and shape of the comet and the distribution of DNA within the comet correlate with the extent of DNA damage.27
DNA Damage Repair
Immediately following radiation-induced DNA damage, a dynamic and well-orchestrated repair process is initiated, which includes DNA damage recognition, chromatin relaxation, formation of multiunit repair protein complexes at sites of DNA damage, DNA DSB repair, and finally repair protein dissolution and chromatin restoration. Despite its complexity, the entire repair process of DNA DSBs is rapid; a majority of base pairs are repaired within 6 hours. Several modalities have been used preclinically to evaluate DNA damage repair, including constant or pulsed-field gel electrophoresis, but these have been criticized because of the need for large radiation doses. The neutral comet assay, which, as described previously, determines initial damage repair, can also be applied to measure DNA DSB repair when evaluated in a time course manner. A methodology that has gained significant popularity in recent years to assess DNA damage repair involves immunofluorescent cytochemistry to assess phosphorylation of H2AX. At DSB sites, the histone H2AX becomes rapidly phosphorylated (γH2AX), forming readily visible foci. The dephosphorylation and dispersal of γH2AX in irradiated cells correlates with repair of the DNA DSBs. Therefore, prolonged expression of γH2AX, for example, when an investigational agent is combined with radiation, suggests abrogation of DNA repair processes. In addition, the γH2AX assay is sensitive enough to detect damage and repair at very low doses in the clinical and subclinical range.28–31 Common methods used to assess DNA damage repair and initial DNA damage are depicted in Fig. 6-3.
Cell Cycle Kinetics
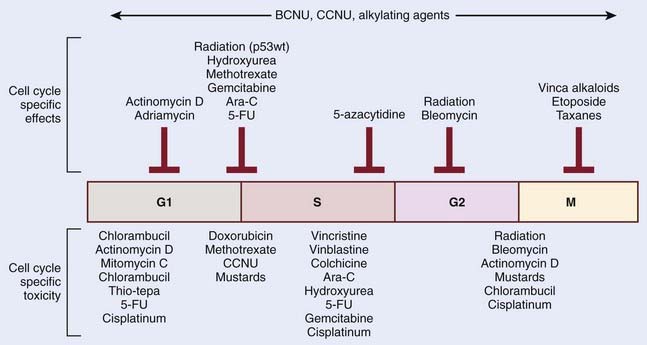
The mechanistic interplay between radiation and chemotherapy through cell cycle kinetics may be appreciated at multiple levels. One potential interaction involves cell cycle redistribution. Focused radiobiologic studies have determined differential radiosensitivity based on cell cycle phase. In general, cells in the G2 or M phases are the most sensitive, and cells in the S phase are the most resistant cells to radiation. This variation in radiosensitivity during the cell cycle can be potentially exploited when designing effective chemoradiation therapy strategies. One example involves the mitotic-spindle poison taxane. These agents act to stabilize microtubules, and thereby prevent chromosome separation at M phase, leading to cell cycle arrest in the radiosensitive G2 and M phases. Another approach by which cell cycle kinetics may be exploited in radiation and chemotherapy interactions involves differential cytotoxicity. Although the S phase of the cell cycle appears to render cells more resistant to the cytotoxic effects radiation, this phase is particularly sensitive to nucleoside analogs that become incorporated in cellular DNA, such as fludarabine or gemcitabine. Therefore, the preferential targeting of cells in radioresistant phases of the cell cycle represents a form of biologic cooperation underlying the observed radioenhancement. An important point to remember is that although chemotherapeutic agents may have a specific cell cycle phase during which they inhibit cell cycle progression, it is often cells in another phase that might be more sensitive. Classic examples are the vinca-alkaloids, which arrest cells in mitosis but are most toxic when cells are exposed in S phase. Fig. 6-4 summarizes these relationships for a series of chemotherapeutic agents.
In addition to cell cycle redistribution, the modulation of checkpoint response represents another approach by which chemotherapeutic agents may influence radiosensitization. The activation of the G2 checkpoint allows for DNA repair before progression into mitosis and is considered to protect against radiation-induced cell death.32 Cell cycle kinetics is typically assessed in vitro by way of flow cytometry. By using a DNA-specific stain (typically propidium iodide or Hoechst) the DNA profile can be determined based on the relative amount of DNA. When relative fluorescence (i.e., the amount of DNA per cell) is plotted as a function of cell number, cells in G1 have the least amount of DNA per cell (2n) and therefore represent the first spike on this plot. Cells accumulate DNA during the S phase, representing the intermediate region, and finally, cells in G2 and M have the most quantitative DNA per cell (4n), and therefore represent the final spike on this plot. However, this approach, which assesses DNA quantity per cell, is unable to differentiate cells in G2 and M phases. Therefore, to distinguish between G2 and mitotic cells, in addition to labeling cellular DNA, cells are also labeled with an antibody specific to phosphorylated histone H3, which is specifically expressed in mitotic cells. Done as a function of time after irradiation, this analysis provides a measure of the progression of G2 cells into M phase and thus the activation of the G2 checkpoint.33
Tumor Microenvironment/Hypoxia
In addition to the aberrant genetic alterations driving uncontrolled tumor growth, the surrounding microenvironment of a tumor plays a critical role in its continued growth and may also influence therapeutic response. An important way in which the microenvironment influences tumor growth is by way of its supporting vasculature. The continued growth of a tumor requires the formation of new blood vessels to facilitate the delivery of nutrients and oxygen, a process called angiogenesis. A critical mediator of tumor angiogenesis is the vascular endothelial growth factor, and targeting this protein using a variety of molecular agents represents a dominant theme in anticancer therapy in nearly all solid tumor types. Despite a robust angiogenic response, the oxygenation concentrations are quite heterogeneous within a tumor, with a gradient of intratumoral oxygenation governed by the diffusion capacity of host vessels. Therefore, regions within a tumor distant from the supporting vasculature develop subpopulations of hypoxic cells. In addition to distance from vasculature, hypoxic cells are also induced from defective vascularization within a tumor, both in the number of blood vessels and vessel function. For example, tumor blood vessels are commonly irregular and tortuous, and have blind ends, arteriovenous shunts, incomplete endothelial linings, and basement membranes, leading to areas of poor oxygenation within a tumor. It has long been suggested that hypoxia contributes to radiation resistance,34 a phenomenon that was initially interpreted as reflecting the requirement for oxygen as a source of radiation-induced free radicals that mediate tumor cell killing. Although mechanisms still remain unclear, a more generally accepted principle is that hypoxia influences radiation response at a molecular level, modulating tumor phenotype and angiogenesis through upregulation of key mediators, including hypoxia-inducible factor (HIF-1).35
Although the evaluation of angiogenesis inhibitors as anticancer agents is apparent, their application when combined with radiation is not as intuitive. For example, based on these proposed mechanisms, although angiogenesis inhibitors purportedly prevent continued growth of the tumor, it may also contribute to inducing hypoxia and theoretically minimizing therapeutic efficacy when combined with radiation. However, tumor vasculature is often functionally and structurally abnormal and contributes to hypoxia through spatial and temporal heterogeneity in tumor blood flow. Recent findings suggest that certain angiogenic agents may normalize the abnormal structure and function of tumor vasculature to abrogate hypoxia and potentially even increase the efficacy of conventional therapies.17
Cell Repopulation
Both radiation therapy and chemotherapy are typically administered in multiple, temporally spaced, doses. With respect to radiation treatments, this is to allow for recovery from sublethal radiation-induced damage and allow repopulation of normal tissues between treatments. However, repopulation of surviving tumor cells can also occur during this interval, increasing tumor burden. In addition, the rate of repopulation may increase during a protracted course of therapy, a phenomenon termed accelerated repopulation, which may further limit the effectiveness of therapy.4 Although studies aimed to quantify accelerated repopulation are often confounded by multiple factors, often being derived from retrospective data with a heterogeneous dose of radiation per fraction, their findings are striking. In an important paper, Withers and colleagues36 analyzed pooled clinical data for TCD50 in squamous cell carcinoma of the head and neck. A marked increase in TCD50 was demonstrated if the treatment lasted more than 4 weeks, which was attributed to accelerated repopulation. Further, the added radiation dose required to overcome repopulation has been estimated to range from 0.5 to 1 Gy per day of prolonged treatment.4 The critical importance of minimizing treatment time in cancer therapy has been corroborated by a large prospective randomized trial that demonstrated clinical gains in head and neck cancer with accelerated fractionated radiotherapy (delivered for 6 weeks) when compared with a conventional fractionated radiotherapy (delivered for 7 weeks).37

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


