Chapter Outline
GLUCOSE PHOSPHATE ISOMERASE DEFICIENCY
PHOSPHOFRUCTOKINASE DEFICIENCY
TRIOSE PHOSPHATE ISOMERASE DEFICIENCY
GLYCERALDEHYDE-3-PHOSPHATE DEHYDROGENASE DEFICIENCY
PHOSPHOGLYCERATE KINASE DEFICIENCY
2,3-BISPHOSPHOGLYCERATE MUTASE DEFICIENCY
MONOPHOSPHOGLYCERATE MUTASE DEFICIENCY
LACTATE DEHYDROGENASE DEFICIENCY
ABNORMALITIES OF ERYTHROCYTE NUCLEOTIDE METABOLISM
The mature erythrocyte, devoid of nucleus, mitochondria, ribosomes, and other organelles, has no capacity for cell replication, protein synthesis, or oxidative phosphorylation. The glycolytic production of adenosine triphosphate (ATP), the sole known energy source of erythrocytes, is sufficient to meet their limited metabolic requirements. The discovery that hemolytic anemia may result from any of several glycolytic enzymopathies has underscored the dependence of erythrocytes on glycolysis. In this chapter the clinical, biochemical, and genetic features associated with abnormalities in erythrocyte glycolysis are described in detail.
Hereditary hemolytic anemias resulting from altered erythrocyte metabolism are distinguished from hereditary spherocytosis by the absence of spherocytes on the peripheral blood smear, by normal osmotic fragility of fresh erythrocytes, by a partial therapeutic response to splenectomy, and by a recessive mode of inheritance. Hemoglobin structure and synthesis are normal. Because no specific morphologic abnormality is associated with these disorders, they have become known as congenital nonspherocytic hemolytic anemias (CNSHAs). Although CNSHAs are usually transmitted in an autosomal recessive fashion, phosphoglycerate kinase (PGK) deficiency is an X-linked abnormality, and adenosine deaminase (ADA) overproduction is an autosomal dominant disorder. Symptoms and signs may be limited to the manifestations of hemolysis or, if the enzymopathy is present in other tissues, may involve other organ systems. The pattern of involvement of nonerythroid tissues may be of assistance in diagnosis.
Initial attempts to classify these anemias were based on the autohemolysis test, in which saline-washed erythrocytes were incubated without glucose in vitro at 37° C under sterile conditions and the percentage of hemolysis was determined after 48 hours. In 1961, Valentine and associates identified a deficiency of erythrocyte pyruvate kinase (PK) in three patients with CNSHA. Subsequently abnormalities of other glycolytic enzymes have also been associated with CNSHA, as indicated in Figure 17-1 . Specific alterations in protein structure underlie many of the enzyme deficiency states, and many of the underlying mutations have been identified.
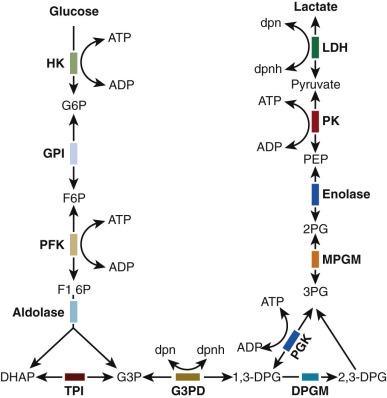
The presence of a glycolytic enzymopathy should be suspected when chronic hemolysis occurs in the absence of marked abnormalities in erythrocyte morphology or osmotic fragility. An exception to the usually unremarkable red cell morphology in CNSHA is the pronounced basophilic stippling found in pyrimidine-5′-nucleotidase (P-5′-N) deficiency. Hemoglobin electrophoresis, stains for inclusion bodies, hemoglobin heat stability, acid hemolysis, and appropriate studies for immune hemolysis are normal. The autohemolysis test, previously used to screen for mutations in the glycolytic pathway, has at best limited value in the evaluation of CNSHA. Unfortunately no other simple, convenient laboratory screening test has been developed that will unequivocally reveal the presence of a glycolytic enzymopathy. Therefore the appropriate diagnostic strategy for the evaluation of a suspected enzymopathy is first to eliminate easily identified causes of hemolysis, such as autoantibodies, hemoglobinopathies, or spherocytosis, before proceeding to tests for enzyme disorders. Definitive diagnosis depends on quantitative assay of the activity of the suspected enzyme or identification of a specific mutation by deoxyribonucleic acid (DNA) analysis. The availability of such assays is limited, but screening tests for deficiencies in PK, triose phosphate isomerase (TPI), and glucose phosphate isomerase (GPI) can be carried out in any well-equipped clinical laboratory. The in vitro properties of mutant enzyme proteins vary, and characterization of such properties has improved understanding of the genetics and pathogenesis of anemias associated with defective glycolytic enzymes ( Table 17-1 ). Measurement of glycolytic intermediates extracted from freshly obtained erythrocytes, although potentially useful, has little clinical applicability other than the measurement of erythrocyte 2,3-bisphosphoglycerate (BPG). In contrast, measurement of intracellular metabolites is the most convenient way to screen for abnormalities in nucleotide metabolism. Red-cell ATP levels are below normal with overproduction of ADA, whereas P-5′-N deficiency is associated with increased concentrations of red-cell ATP and reduced levels of glutathione. The apparent increase in ATP is in fact the result of the presence of large amounts of cytidine and uridine nucleotides, which are also measured in the enzymatic assay for ATP. Spectral analysis of a deproteinized extract of red cells provides a straightforward means of identifying such nucleotides, and it is a simple method to screen patients for suspected P-5′-N deficiency.
| V max | Maximal enzyme velocity obtainable with saturating substrate concentrations |
| K m | The substrate concentration yielding half-maximal activity; an index of catalytic efficiency |
| pH optimum | The pH at which maximal enzyme activity is present |
| Heat stability | Resistance of enzyme protein to heat denaturation |
| Electrophoretic mobility | Migration of enzyme protein in an electric field |
| Specific activity | Enzyme activity per defined amount of enzyme protein (e.g., milligram); enzyme protein is measured immunologically with antienzyme antibodies |
Caution is needed in interpreting the results of quantitative assays of enzyme activity. First, only surviving cells are available for sampling in circulating blood, and the metabolic circumstances of these favored cells cannot necessarily be extrapolated to indicate the status of cells already hemolyzed. Second, assay in vitro under optimal conditions may not adequately reflect the performance of an enzyme under less favorable circumstances in vivo. Third, the high specific activity of certain enzymes in leukocytes may result in spurious normal values for erythrocyte enzyme activity unless contaminating leukocytes are removed before assay or their contribution to total activity is compensated for by appropriate calculations. Fourth, transfusion therapy with normal erythrocytes within several months before assay may obscure the presence of an enzyme defect. Fifth, the mean enzyme activity that is determined fails to portray the distribution of activity within individual erythrocytes. The endowment of intracellular enzymes is fixed, and protein synthetic ability disappears at the reticulocyte stage; thereafter the inevitable denaturation of enzyme protein that accompanies cell aging reduces enzymatic activity at a rate characteristic for each enzyme. Therefore transient accentuation of reticulocytosis is often accompanied by rising mean enzyme activity. Certain glycolytic enzymes (notably hexokinase [HK] and PK) are strikingly more active in reticulocytes than in postreticulocyte red cells, and the majority of this excess activity is rapidly lost coincident with reticulocyte maturation. The true magnitude of an enzyme deficiency may not be apparent unless comparison is made to blood that is equally rich in reticulocytes or corrections are applied that eliminate the contribution of the reticulocyte subfraction to total enzyme activity.
Finally there is evidence that reversible binding of glyceraldehyde-3-phosphate dehydrogenase (G3PD), HK, and phosphofructokinase (PFK) to the band 3 membrane protein is involved in the regulation of glycolysis, and altered binding of mutant forms of these enzymes (and perhaps others) is not assessed in conventional assays performed on hemolysates.
Hexokinase Deficiency
Clinical Manifestations
CNSHA has been attributed to deficient erythrocyte HK activity in 24 patients ( Table 17-2 ). Severely affected individuals may exhibit neonatal hyperbilirubinemia and thereafter require transfusion at regular intervals for intractable anemia. In patients with mild disease, the hemolysis is fully compensated for and anemia is absent. However, jaundice, reticulocytosis, and splenomegaly are usually present in such patients. Gallstones may be evident, even in early childhood. Hyperhemolytic episodes are not a feature of the disorder. The results of red cell morphologic examination are usually unremarkable, but occasional burr cells, target cells, stippled cells, and densely stained spiculated cells may be observed after splenectomy. The osmotic fragility of fresh erythrocytes is normal, but a fragile population of cells may appear after incubation at 37° C.
| CLINICAL FEATURES | PROPERTIES OF RBC HEXOKINASE | ||||||
|---|---|---|---|---|---|---|---|
| Activity | Kinetic Abnormalities | Stability | Mobility | ||||
| Reference | Inheritance | Anemia | Other | ||||
| — | + | Congenital malformations | 13-24 * | 0 | — | — | |
| Recessive | ++ | 15-20 * | + | Normal | Abnormal | ||
| Recessive | ++ | 16 * | 0 | — | Abnormal | ||
| Recessive | +++ | Hydrops fetalis | 17 | ||||
| Recessive | + | 20 * | 0 | Normal | Normal | ||
| Recessive | ++ | Low platelet and fibroblast HK activity | 20 * | 0 | Low | Normal | |
| Recessive | ++ | Low platelet HK activity | 25 * | + | Normal | Abnormal | |
| Recessive | + | 25 * | 0 | Low | Normal | ||
| Dominant | + | Spherocytes, ovalocytes | 30 * | 0 | Low | Normal | |
| Recessive | + | Psychomotor retardation | 45 † | + | Normal | Normal | |
| Recessive | + | 50 * | 0 | Normal | Normal | ||
| — | + | Congenital malformations | 33 * | + | — | — | |
| Recessive | + | 40-53 * | + | Low | Normal | ||
| — | + | 50 * | + | — | — | ||
| 53 | |||||||
| Dominant | + | 45-91 † | + | Normal | Abnormal | ||
| Dominant | ++ | WBC HK activity low | 75 * | + | Normal | Abnormal | |
| Recessive | ± | 77 † | |||||
* Maximal enzyme activity (V max ) compared with reticulocytosis controls.
† Maximal enzyme activity (V max ) compared with normal red cells.
Biochemistry
In human red cells, HK is a monomer (relative molecular mass: 112,000). It is encoded by a gene ( HK1 ) located on chromosome 10q22. HK activity declines as red cells age. Loss of activity is particularly striking during reticulocyte maturation. In human reticulocytes, two major isoenzymes of HK have been identified by chromatographic techniques. One (HKR) has an apparent half-life in vivo of only 10 days, whereas the other (HK1) has a longer half-life of 66 days. These two proteins are the products of two closely similar messenger ribonucleic acids (mRNAs) that are transcribed from a single gene ( HK1 ) by the use of alternate promoters. Exon 1 is unique to each mRNA species, but the remaining 17 exons are identical. Differential loss of these two isoenzymes appears to explain the biphasic character of the decay in HK activity during erythrocyte aging. An ATP- and ubiquitin-dependent proteolytic system capable of degrading about 80% of HK activity may explain the rapid loss of HK in rabbit reticulocytes, or the loss may be secondary to an intrinsic property of the HK molecule itself. Previous oxidative injury appears to be necessary for recognition and destruction of HK by the ubiquitin-dependent system. Because ATP- and ubiquitin-dependent proteolysis is limited to reticulocytes, it cannot be responsible for the loss of HK in aging human red cells.
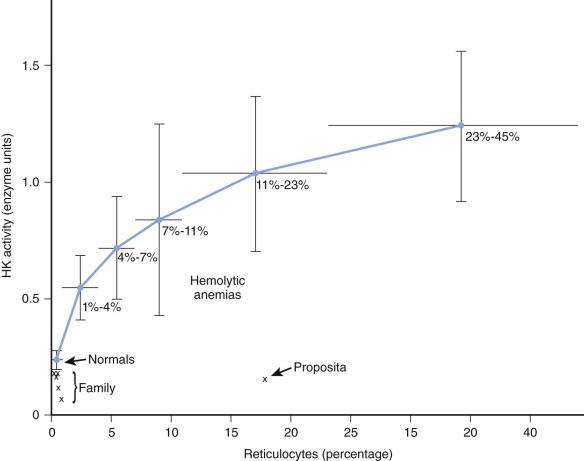
HK is the glycolytic enzyme with the lowest activity in normal red cells, and a variety of observations indicate that it plays a rate-limiting role in erythrocyte glycolysis. The maximal activity of erythrocyte HK from deficient patients has varied from 13% to 91% of normal (see Table 17-2 ). In evaluating these findings, comparisons of enzyme activity between red cell populations of equivalent age must be made. In the case described by Valentine and associates ( Fig. 17-2 ), although HK activity was 62% of the normal value for mature erythrocytes, it was only 14% of the activity found in blood with high reticulocyte counts. Separation of young and old red cell populations by centrifugation revealed only the expected moderate diminution (to 0.11 mol/min/10 10 red blood cells) of HK activity in older cells from this patient. HK activity was even lower (0.075 mol/min/10 10 red blood cells) in an asymptomatic brother, yet no evidence of undue hemolysis was present. However, Figure 17-2 shows that the brother’s cells are actually far less deficient with respect to cell age than the immature cells of the propositus. The impact of HK deficiency is clearly greater in energetic young erythrocytes, whereas cells that survive to an older age can meet their limited metabolic requirements even at very low HK levels. As the erythrocyte ages, in vivo changes in stability or kinetics peculiar to mutant HK may also render older cells liable to undergo premature hemolysis. In rats and rabbits, HK from immature erythrocytes has a higher Michaelis constant ( K m ) for glucose than mature cells do, but such is not the case in normal human erythrocytes.
In keeping with their enzymatic defect, HK-deficient erythrocytes have usually demonstrated subnormal glucose consumption and lactate production in vivo. Such cells also metabolize fructose poorly but utilize mannose or galactose normally, because these substrates are not metabolized by HK. Some HK-deficient erythrocytes are capable of normal glucose consumption at the glucose concentrations (5 mmol/L) customarily found in plasma but utilize glucose poorly or not at all at lower glucose concentrations, either because of an abnormally low affinity for glucose or because of enzyme instability under conditions of low substrate availability. Such erythrocytes may encounter a particularly unfavorable metabolic environment within the spleen. The concentration of glucose in normal splenic homogenates has been found by Necheles and coworkers to be only 5 to 11 mmol/g of tissue, and in a patient with HK deficiency the concentration was even lower (1.1 mmol/g of tissue). Furthermore, because splenic tissue metabolizes glucose rapidly, prolonged vascular pooling in the spleen is probably accompanied by profound local hypoglycemia. Erythrocytes with unfavorable kinetics for glucose metabolism are clearly at a disadvantage in competing with reticuloendothelial cells for such a reduced glucose supply. Another disadvantage of the splenic environment is its relative acidity. The optimal pH of erythrocyte HK is approximately 8; at lower pH values, diminished enzyme activity may be expected. Possibly of greater importance, at low pH values, glucose 6-phosphate, a potent inhibitor of HK, accumulates because of PFK inhibition. An erythrocyte that has diminished HK activity under optimal pH conditions will be further compromised in the acidic environment of the spleen. The clinical improvement that follows splenectomy attests to the importance of this organ in the pathogenesis of hemolysis. No splenic sequestration of chromated autologous erythrocytes was noted in two patients. Thus in this disorder the red cells may be damaged by the spleen and die elsewhere.
Significant alterations in intracellular metabolites are associated with defective HK function. The erythrocyte ATP concentration is sometimes, but not always subnormal. The glucose-6-phosphate concentration is reduced to approximately half normal, and the concentrations of other more distal intermediates, most notably 2,3-BPG, are also usually reduced. These metabolites may exert a significant regulatory influence on glycolysis. For example, Brewer has shown that concentrations of 2,3-BPG in the physiologic range inhibit HK, so the low 2,3-BPG levels in HK-deficient red cells may facilitate the performance of available HK. The increased affinity of hemoglobin for oxygen expected to be associated with subnormal 2,3-BPG levels has been documented in one patient with anemia and HK deficiency by Delivoria-Papadopoulos and coworkers and by Oski and associates. This patient, whose hemoglobin–oxygen affinity (P50) was 19 mm Hg (normal, 27 ± 1.2), was capable of just minimal exercise despite only moderate anemia (hemoglobin, 9.8 g/dL). On exercise, her central venous partial pressure of oxygen (pO 2 ) promptly fell to minimal levels as oxygen consumption rose. Increased oxygen delivery was achieved primarily by an increase in cardiac output, because the unfavorable oxygen affinity curve precluded any substantial further desaturation of hemoglobin. Thus the altered concentration of intracellular metabolites induced by HK deficiency may, as in this patient, accentuate symptoms associated with anemia.
Because of its reactive sulfhydryl group, HK is susceptible to oxidant inactivation in the absence of sufficient glutathione. Both normal and low glutathione levels have been reported in HK deficiency, but Heinz bodies have not been observed. Resting hexose monophosphate (HMP) shunt activity, measured with glucose-1 (C 14 ), was quantitatively normal in one patient despite subnormal glucose consumption, but stimulation with methylene blue was suboptimal. Failure of the shunt at low glucose concentrations has also been noted in a patient with HK mutancy with a high K m for glucose. Methylene blue–stimulated methemoglobin reduction was subnormal in the single instance that it was evaluated. When HK-inactivating antibodies are incorporated into normal red cells by the process of hypotonic lysis and isotonic reannealing, enzyme-deficient cells exhibit a greatly impaired response to HMP shunt stimulation by methylene blue. These studies, though not indicating a central role for defective shunt activity in the pathogenesis of hemolysis, do suggest that under unusual circumstances, HK-deficient cells might be compromised by a limited shunt. Such circumstances might arise on exposure to a potent oxidant in the low glucose environment of the spleen.
Genetics
Inheritance of HK deficiency is autosomal recessive. Biochemical identification of asymptomatic carriers is not always possible, because enzyme activity often falls within the low-normal range. In a few pedigrees (see Table 17-2 ), the heterozygous state appears to be severe enough to result in hemolytic anemia. One such heterozygote appeared to be doubly heterozygous for HK and glucose-6-phosphate dehydrogenase (G6PD) deficiency.
The qualitative abnormalities characteristic of mutant HK (see Table 17-2 ) may reflect either a structural or a regulatory gene mutation. On electrophoresis, mutant HK lacks one or more of the normal bands of activity, but no bands migrating in an abnormal position have been observed. The various bands represent the presence of several isozymes of HK. Diminished synthesis of one or more isozymes with predominance of the remaining isozymes accounts for the electrophoretic differences observed (see Table 17-2 ). DNA analysis has defined two separate HK mutations in a compound heterozygote who exhibited nonspherocytic hemolytic anemia. One was a missense mutation ( 1667T → C ) and the other was a deletion of exon 5 (96 base pairs [bp]). Expression of each HK mutation in a bacterial system allowed recovery of sufficient purified enzyme to determine that the missense mutation completely abolished HK activity, whereas the deletion reduced activity to about 10% of normal. In another individual, a homozygous missense mutation in the active site of HK1 ( C2039 → G in exon 15) was discovered. In a Japanese family, a stillborn fetus with HK deficiency was found to be homozygous for a large deletion that removed exons 5 through 8 in HK mRNA and led to premature termination of translation. A mutation in the erythroid-specific promoter of HK-1 inherited along with a missense mutation in exon 3 that caused aberrant splicing of both HK-R and HK-1 led to HK deficiency and mild chronic hemolytic anemia. The kinetic abnormalities noted in many other mutant forms of HK undoubtedly also reflect as yet undefined structural abnormalities in enzyme protein. *
* References
Studies of the tissue distribution of HK deficiency have been performed in blood cells and cultured fibroblasts. Electrophoresis of leukocyte or platelet HK reveals an anodal isozyme (HK-3) distinct from those of the erythrocyte (HK-1 and HK-2), as well as a shared isozyme (HK-1). Leukocyte HK activity has been normal in some patients, but the qualitative abnormality in leukocyte HK described by Necheles and colleagues, as well as the case of generalized HK deficiency in all blood cells reported by Rijksen and coworkers, indicates that to some extent the enzyme is under common genetic control in different tissues. When platelet HK activity has been low, platelet function has been normal despite subtle defects in in vitro energy metabolism. Cultured fibroblasts from two individuals with different HK mutants contained HK with properties and activity like those found in red cells, thus suggesting that this source of fetal tissue could be used for prenatal diagnosis.A murine model of HK deficiency, termed Downeast anemia, is caused by homozygous inheritance of a mutation that markedly decreases the expression of HK-1 in erythroid tissues, spleen, and kidney. The mutation is the result of insertion of a transposon into intron 4 of the HK1 gene. Mice exhibit nonspherocytic hemolytic anemia, marked reticulocytosis, splenomegaly, and striking accumulation of iron in the liver and kidney. Echinocytes and red cells split into two poles of hemoglobin connected only by thin strands of membrane are features of erythrocyte morphology in affected homozygous mice that are of interest because they have not been reported in human HK deficiency.
Therapy
Treatment consists of red cell transfusion as indicated, supplemental folic acid, and close observation for cholelithiasis. Splenectomy may alleviate but does not eliminate the anemia. †
† References .
Glucose Phosphate Isomerase Deficiency
Clinical Manifestations
A deficiency of erythrocyte GPI has been reported in more than 60 patients with congenital hemolytic anemia. Hemolytic anemia usually appears in infancy and often requires red cell transfusion therapy. Hyperbilirubinemia, hydrops fetalis, or death may occur during the neonatal period. Several patients have experienced hyperhemolytic crises after infections or drug exposure. In some of these individuals G6PD, as well as GPI, has been deficient. Hemolytic anemia is usually the sole clinical manifestation of GPI deficiency. A subset of patients also exhibit neurologic dysfunction. In one patient, neuromuscular abnormalities were correlated with a severe reduction in muscle and cerebrospinal fluid GPI activity. Blockade of glycolysis at the GPI step may divert the flow of glucose metabolism in the direction of glycogen synthesis. Several patients with GPI deficiency have been reported to have increased hepatic glycogen stores, and in one, muscular fatigue was severe enough to suggest a diagnosis of glycogen storage disease. Animal models of GPI deficiency and chronic hemolytic anemia have been developed in the mouse. The clinical and biochemical features of the disease in mice closely resemble those found in humans.
Red cell morphologic characteristics are generally similar to those seen in other types of CNSHA. In patients with severe anemia, dense, spiculated, or “whiskered” microspherocytes have been noted after splenectomy. In one instance, sufficient numbers of such cells were present before splenectomy to suggest the diagnosis of hereditary spherocytosis. In another, the predominant morphologic abnormality was stomatocytosis. The reticulocytosis may be profound. Mean corpuscular volume is elevated (97 to 139 fL). With incubation at 37° C, a variable fraction of erythrocytes may exhibit abnormally increased osmotic fragility, whereas fresh cells are usually normal. Survival of chromated autologous red cells is reduced, often but not invariably, with evidence of splenic sequestration.
Biochemistry
GPI is a dimer composed of two identical subunits, each with a relative molecular mass of approximately 60,000. Subunit synthesis is directed by a single genetic locus on chromosome 19. The crystal structure of human GPI has been defined. GPI is a multifunctional protein. The dimeric form exhibits glycolytic enzyme activity, whereas the monomeric form functions as a cytokine. Monomeric GPI is 100% homologous to neuroleukin, a neurotrophic growth factor secreted by lectin-stimulated T cells ; to autocrine motility factor, which stimulates cell motility and the growth of metastases ; and to a differentiation and maturation mediator for human myeloid leukemia cells in tissue culture. Altered neuroleukin function may explain the neurologic disorder associated with several GPI mutations. Alternatively, induced GPI mutations in Chinese hamster ovary cell lines have an adverse effect on glycerolipid biosynthesis, suggesting that deficient GPI activity affects important non-glycolytic pathways.
The metabolic events that precede hemolysis of GPI-deficient erythrocytes are poorly understood. Erythrocyte glycolysis is impaired in vivo, as reflected by an increased ratio of substrate to product (i.e., glucose 6-phosphate to fructose 6-phosphate) in freshly obtained GPI-deficient cells. Paradoxically, with only occasional exceptions, such cells are fully capable of glycolysis in vitro. In contrast to HK deficiency, in which 2,3-BPG levels are low, sufficient glycolysis generally occurs in GPI deficiency to maintain the 2,3-BPG concentration at or above the normal level. Except for three patients with diminished ATP concentrations, two of whom also exhibited reduced in vitro glycolysis for cell age, the erythrocyte ATP concentration has been normal.
A profound defect in recycling of fructose-6-phosphate through the pentose phosphate pathway has been observed repeatedly in GPI-deficient red cells. The increased formation of Heinz bodies and glutathione instability after exposure to acetylphenylhydrazine, an abnormal ascorbate cyanide test, and diminished concentrations of red cell glutathione in fresh erythrocytes all suggest that diminished shunt activity in vivo may contribute to hemolysis.
With rare exceptions, mutant forms of red cell GPI exhibit considerable thermal lability in vitro, thus making it likely that enzyme lability in vivo as red cells age will lead to premature metabolic collapse and hemolysis. Separation of red cells by centrifugation into young and old subpopulations has usually demonstrated accelerated decay of enzyme activity in the older cells. Arnold and coworkers simulated the in vivo process of aging by incubating red cells in vitro at 37° C for 8 days and changing the incubation medium often to ensure that glucose availability and pH remained constant. GPI activity declined by 66% in GPI-deficient red cells during incubation, with a level of only 6% of normal being reached, and lactate production, normal at the onset, was reduced to 11% of normal. In contrast, normal reticulocyte-rich blood lost only 6% of the original GPI activity after 8 days, and lactate production fell only 7%. If mannose rather than glucose was used, GPI-deficient and normal red cells made equivalent amounts of lactate, and there was little or no loss in lactate production after 8 days. The normal glycolytic rate noted with mannose, which is isomerized by mannose phosphate isomerase and thus bypasses the GPI reaction, clearly pinpoints defective GPI activity as the cause of glycolytic failure in GPI-deficient cells. ATP depletion with consequent erythrocyte rigidity and reticuloendothelial entrapment would be anticipated to follow failure of glycolysis. GPI-deficient red cells are indeed less deformable than normal cells, particularly when comparison is made to young, reticulocyte-rich populations of cells. Goulding studied a 9-year-old patient with GPI deficiency and priapism and concluded that the priapism was the consequence of abnormal erythrocyte deformability. Studies by Coetzer and Zail revealed aggregation of membrane spectrin in GPI-deficient erythrocytes. The extent of the aggregation is a function of cell age.
Even reticulocytes may be severely GPI deficient. Deficient reticulocytes with limited anaerobic glycolytic capability may also be incapable of effective oxidative phosphorylation in the acidic, hypoglycemic splenic environment (see Pyruvate Kinase Deficiency ), thereby leading to metabolic failure and hemolysis. Large numbers of reticulocytes were found when specimens from the spleen of a patient with GPI deficiency were examined by transmission electron microscopy. Furthermore, the reticulocyte count often increases after splenectomy. Because hemoglobin levels also increase, this observation suggests survival of a population of reticulocytes that would otherwise be hemolyzed almost immediately after their release from bone marrow.
Genetics and Inheritance
Like most other glycolytic enzymopathies, GPI deficiency is inherited as an autosomal recessive trait. Heterozygotes are hematologically normal but exhibit reduced erythrocyte GPI activity (usually to about 50% of normal). They inherit one mutant and one normal GPI allele, which results in the synthesis of two unlike GPI subunits that may combine in one of three ways to form a normal homodimer, a mutant homodimer, or a heterodimer that contains both normal and mutant subunits. Electrophoresis of GPI from the erythrocytes of heterozygotes demonstrates one to three bands depending on the extent to which the charge or activity of the mutant subunit is altered. Posttranslational events, such as oxidation of enzyme protein, may alter the electrophoretic pattern and confuse its interpretation.
GPI mutations associated with hemolytic anemia are listed in Table 17-3 . Of the 31 known mutations, 2 are splice site, 3 are nonsense, and 31 are missense mutations involving a single amino acid substitution. The mutations are found in 12 of the 18 exons and are generally unique to one or, at most, a few families. The rarity of GPI deficiency and the heterogeneity of the mutations identified indicate that no selective advantage is conferred to affected individuals. Half of the homozygotes or compound heterozygotes for missense mutations have mild to moderate hemolytic anemia, and half have severe anemia. In contrast, all compound heterozygotes for missense mutations and either splice site or nonsense mutations have severe hemolytic anemia. Hemolytic anemia occurs in individuals when GPI activity drops below about 40% of the normal mean activity. Substrate kinetics and the optimal pH of mutant enzymes have almost always been normal, but most mutant enzymes have exhibited varying degrees of thermal instability. Study of recombinant GPI enzyme protein generated from 16 different human mutations has clarified the relation between specific mutations, their location within the three-dimensional structure of GPI, and enzyme properties (catalytic activity, enzyme kinetics, and thermostability). Mutations at or near the active site have the most severe consequences, whereas areas more distant generally have lesser effects. Thermostability is affected by different mutations to varying degrees. Short of direct detection of the mutation at the DNA level, the most useful means of classifying the numerous GPI variants reported has been on the basis of stability, electrophoretic mobility, or residual enzyme activity in red cells and leukocytes. ‡ Until DNA mutation analysis has been completed on these variants, it will not be clear how many are truly unique. Immunologic titration of functionally inactive enzyme protein in red cells indicates that some mutant GPI alleles are “silent” and produce no detectable enzyme protein, whereas others produce structurally altered protein with varying degrees of activity and stability in vivo.
| Variant | Ethnic Origin | Genotype | Exon | Amino Acid Change | Red Cell GPI Activity * |
|---|---|---|---|---|---|
| Homozygotes: Missense Mutations | |||||
| Matsumoto | Japanese | 14C → T | 1 | Thr5Ile | 27 |
| Unnamed | American Indian | 247C → T | 3 | Arg83Trp | 16.4 |
| Sarsina | Italian | 301G → A | 4 | Val101Met | 7.2 † |
| Iwate | Japanese | 671C → T | 7 | Thr224Met | 10.2 |
| Unnamed | Turkish | 970G → A | 12 | Gly324Ser | 49.7 † |
| Morcone | Italian | 1028A → G | 12 | Gln343Arg | 7.2 † |
| Narita | Japanese | 1028A → G | 12 | Gln343Arg | 32.4 |
| Mount Scopus | Ashkenazi | 1039G → A | 12 | Arg347Cys | 14.8 † |
| Unnamed | Spanish ‡ | 1040G → A | 12 | Arg346Hist | 18 † |
| Unnamed | Hispanic | 1415G → A | 16 | Arg472His | 15.4 |
| Unnamed | Turkish | 1415G → A | 16 | Arg472His | 24.9 |
| Unnamed | Indian ‡ | 1459C → T | 16 | Leu487Phe | 8.6 |
| Unnamed | ? English | 1574T → C | 18 | Ile525Thr | 3 † |
| Fukuoka | Japanese | 1615G → A | 18 | Asp539Asn | 6.4 |
| Compound Heterozygotes: Nonsense/Missense Mutations | |||||
| Stuttgart | German | 43C → T/1028A → G | 1, 12 | Gln15Stop/Gln343Arg | 27.9 † |
| Elyria | Caucasian | 223A → G/286C → T | 3, 4 | Arg75Gly/Arg96Stop | 4 † |
| Bari | Italian | 286C → T/584C → T | 4, 6 | Arg96Stop/Thr196Ile | 14.9 † |
| Unnamed | Russian | 286C → T/1039C → T | 4, 12 | Arg96Stop/Arg347Cys | 19 |
| Zwickau | German | 1039C → T/1538G → A | 12, 17 | Arg347Cys/Trp513Stop | 21 † |
| Catalonia | Spanish ‡ | 1648A → G | 18 | Lys550Glu | <7 † |
| Compound Heterozygotes: Splice Site/Missense Mutations | |||||
| Mola | Italian | 584C → T /del1473-IVS16(+2) | 6, 16 | Thr195Ile/splice site | 12.9 † |
| Nordhom | German | 1028A → G / IVS15(−2)A → C | 12/IVS15 | Gln343Arg/splice site | 11.4 † |
| Compound Heterozygotes: Missense/Missense Mutations | |||||
| Homburg | German | 59A → C / 1016T → C | 1, 12 | His20Pro/Leu339Pro | 3.5 † |
| Barcelona | Spanish ‡ | 341A → T / 663T → G | 4, 7 | Asp113Val/Asn220Lys | 20 |
| Unnamed | ? English | 475G → A / 1040G → A | 5, 12 | Gly159Ser/Arg347His | 6 † |
| Unnamed | Black American | 671C → T / 1483G → A | 7, 17 | Thr224Met/Glu495Lys | 12.2 |
| Unnamed | Black American | 818G → A / 1039C → T | 10, 12 | Arg273His/Arg347Cys | 15.9 |
| Unnamed | Caucasion | 833C → T / 1459C → T | 10, 16 | Ser278Leu/Leu487Phe | 9.8 |
| Unnamed | Hispanic | 898G → C / 1039C → T | 11, 12 | Ala300Pro/Arg347Cys | 25.4 † |
| Kinki | Japanese | 1124C → G / 1615G → A | 13, 18 | Thr375Arg/Asp539Asn | 3.7 † |
| Calden | German | 1166A → G / 1549 → G | 13, 18 | His389Arg/Leu517Val | 12 † |
* International units per gram of hemoglobin.
† Severe disease (splenectomy, frequent red cell transfusions, and/or reticulocyte count >15%).
‡ The Spanish patients are described by Repiso et al and the Indian patient by Warang et al.
‡ References .
A single GPI isozyme is present in all human tissues. GPI deficiency is usually less severe in nonerythroid tissues than in erythrocytes because nonerythrocytic tissues retain the ability to synthesize GPI subunits. Clinical abnormalities outside the hematopoietic system are rare and, if present, are neuromuscular in nature. Leukocytes are capable of normal phagocytosis and chemotaxis despite a reduction in GPI activity to 25% to 73% of normal, but if GPI activity is more severely depressed, granulocyte function is impaired. Similarly, platelet GPI may be only 20% to 30% of normal; however, clot formation, platelet aggregation, and other clotting studies are normal. Prenatal diagnosis is feasible by DNA analysis if the mutations are known or by measurement of GPI activity in amniotic fluid fibroblasts or chorionic villus trophoblasts.Therapy
Transfusion requirements are usually eliminated by removal of the spleen, but the anemia persists. The postsplenectomy hemoglobin levels of 6.7 to 10.3 g/dL and reticulocyte counts of 36% to 73% observed in three siblings by Paglia and coworkers reflect the magnitude of the continued hemolysis that may be present. Attempts by Arnold and colleagues to enhance glycolysis in a GPI-deficient patient by intravenous administration of methylene blue or inorganic phosphate (P i ) did not have a lasting benefit.
Phosphofructokinase Deficiency
Clinical Manifestations
Inherited deficiency of PFK can involve erythrocytes, muscle, or both, depending on the PFK subunit affected and the nature of the biochemical defect ( Table 17-4 ). Although low erythrocyte PFK activity and mild hemolytic anemia are commonly found in type VII glycogen storage disease (Tarui disease), the dominant clinical feature of this disorder is exertional myopathy caused by deficient muscle PFK activity. Physical activity is limited not by anemia but by weakness, easy fatigability, and severe muscle cramps associated with the myopathy. The disease may be evident at birth and cause death during infancy as a result of respiratory insufficiency and other complications, or it may be so mild that it is not manifested until old age. However, in most affected individuals the disorder is first detected during adolescence or young adulthood. The diagnosis may be suspected if no lactate is produced during an ischemic (anaerobic) forearm exercise test, but confirmation requires muscle biopsy for determination of PFK activity or noninvasive magnetic resonance imaging (MRI) studies of muscle carbohydrate metabolism.
| RED BLOOD CELL | MUSCLE | ||||||
|---|---|---|---|---|---|---|---|
| Type | Patients | Affected PFK Subunit | Hemolysis | PFK * | Myopathy | PFK * | Other |
| I | 18 | M (absent or unstable) | Present | 29-64 | Present | 0-5 | Hyperuricemia, arthritis |
| II | 3 | NA | NA | 17 | Present | 0-6 | |
| III | 3 | M (unstable) | Present | 8-62 | Absent | 100 | |
| IVa | 2 | M (unstable) | Absent | 28-50 | Absent | 78 † | Asymptomatic |
| IVb | 3 | L (unstable) | Absent | 60-65 | Absent | NA | Asymptomatic |
| V | 3 | NA | NA | 75 † | Present | 2-6 | Arthritis |
When PFK deficiency is confined to erythrocytes, there are no symptoms of myopathy, and the blood lactate response to anoxic exercise is normal. Such patients may be hematologically normal or exhibit mild to moderate hemolytic anemia. In general, red cell morphologic characteristics are not strikingly abnormal, although prominent basophilic stippling has occasionally been noted.
Biochemistry
PFK is one of several glycolytic enzymes that reversibly bind to the inner aspect of the erythrocyte membrane. Binding, which is thought to occur between the aminoterminal position of the transmembrane protein band 3 and the adenine nucleotide binding site, located in a cleft between the two dimers that form the PFK tetramer, may serve to both activate the enzyme and protect it against proteolytic degradation during erythrocyte aging.
The active form of human erythrocyte PFK is a tetramer (relative molecular mass: 380,000) composed in varying combinations of two different subunits, one identical to the M subunit found in muscle PFK and the other identical to the L subunit found in liver PFK. The relative molecular mass of the M subunit is 85,000, and that of the L subunit is 80,000. About 50% of the erythrocyte enzyme is formed from M subunits, whereas muscle PFK is composed entirely of M subunits. A deficiency in M subunits severely depresses muscle PFK activity and results in myopathy, but it has a lesser effect on erythrocyte PFK because residual L subunits, under separate genetic control, combine to form an active L4 tetramer of PFK. However, PFK formed entirely from L subunits is unstable to heat or dilution in vitro and is more sensitive to ATP inhibition than muscle (M4) PFK. At the in vivo concentrations of ATP that are present within normal erythrocytes, the enzyme activity of L4 PFK tetramers is severely inhibited, thus possibly explaining the presence of hemolytic anemia even when enzyme activity, as measured in vitro, is approximately 50% of normal. Mice that lack the M subunit but retain the L subunit have a disease similar to Tarui disease in humans and exhibit chronic hemolysis. Most recognized examples of PFK deficiency are the result of either missing or structurally altered M subunits. An interesting exception was found in a clinically normal individual, fortuitously discovered when he volunteered to serve as a “control” during studies of red cell PFK carried out by Vora and colleagues. Normal M subunits but mutant, unstable L subunits were found in his red cells. There was no myopathy, and although erythrocyte PFK was only 65% of normal, hemolytic anemia was not present, presumably because residual enzyme activity within the red cell was entirely the result of the presence of M4 tetramers of PFK. The relatively greater stability and lesser susceptibility to ATP inhibition of this form of PFK apparently allowed adequate enzyme activity under conditions normally found within the red cell in vivo.
Because of the central role of PFK in regulation of erythrocyte metabolism, it is not surprising to find that a deficiency of this important enzyme is associated with hemolysis, but the mechanism of hemolysis is not well understood. Erythrocyte sodium and potassium concentrations, sodium influx, and lactate production were normal in one patient. Despite their normal glycolytic capability in vitro, deficient cells were incapable of maintaining normal ATP concentrations in vivo. The low (73% of normal) intracellular ATP concentration in these cells, though indicative of an abnormality in cellular metabolism, may also exert a positive influence by partially relieving the inhibitory influence of ATP on PFK. Extensive in vitro study of erythrocytes obtained from a Swedish family with Tarui disease and mild compensated hemolytic anemia found elevated intraerythrocytic calcium levels associated with increased calcium permeability of the erythrocyte membrane. High intracellular calcium levels activated the Gardos channel with subsequent ATP depletion, potassium depletion, increased mean corpuscular hemoglobin concentration (MCHC), and diminished erythrocyte deformability. The authors point out that the PFK and Ca 2+ channel genes are in close proximity to one another on chromosome 12q, but the reason for abnormal Ca homeostasis remains undefined.
The complex interaction between metabolites that may dictate actual PFK activity in vivo is illustrated by physiologic studies performed on four individuals with Tarui disease. At usual levels of physical activity, the pattern of erythrocyte glycolytic intermediates clearly reflected inhibition at the PFK step, and the concentration of the important downstream metabolite 2,3-BPG was only 50% of the level found in normal red cells. After the patients had 2 days of bed rest, their red cell 2,3-BPG levels sank to just one third of normal. Subsequently ergometric exercise on a bicycle eliminated the glycolytic intermediate pattern of PFK inhibition and allowed downstream intermediates, including 2,3-BPG, to increase toward normal. Release of large amounts of inosine and ammonia from enzymopathic muscle during exercise into plasma was observed in these patients. Ammonia is a powerful activator of PFK; inosine can be metabolized to lactate by glycolytic pathways (i.e., the HMP shunt) that bypass the PFK reaction. Thus the muscle metabolic abnormalities created by PFK deficiency generate metabolites that alleviated the enzymopathy in erythrocytes. (Diversion of the flow of erythrocyte glycolysis through the HMP shunt by the block at PFK may also generate increased amounts of purines and pyrimidines from 5-phosphoribosylpyrophosphate [PRPP].) The hyperuricemia sometimes noted in individuals with PFK deficiency (see Table 17-4 ) may be explained on this basis.
An inherited deficiency of PFK found in English springer spaniels, whippets, and wachtelhunds allows interesting comparisons to be made with the human condition. As in humans, canine PFK deficiency is an autosomal recessive disorder. Red cell PFK levels are only 7% to 22% of normal in homozygotes because the muscle subunit, which is lacking, accounts for the majority of the available subunits in normal dog red cells. PFK deficiency in dogs is associated with severe hemolytic anemia. Newborn dogs are not anemic, because there is a greater abundance of L subunits and thus of functional PFK enzymes in their red cells. Hemolytic anemia appears because the normal developmental pattern of replacement of L by M subunit synthesis occurs in a setting in which M subunits either are not synthesized or are defective.
A unique feature of PFK deficiency in dogs is episodic hemolysis induced by hyperventilation during exercise, mating, barking, or other similar activities. Dog red cells with high sodium levels exhibit spontaneous hemolysis at alkaline pH; even the small pH change induced by hyperventilation is sufficient to generate the effect in PFK-deficient animals. Underlying the susceptibility of PFK-deficient dog red cells to hyperventilation-induced hemolysis may be their low 2,3-BPG levels, which increase intracellular pH. Raising 2,3-BPG levels to normal in vitro normalizes the response to alkalinity. In comparison to humans, there is less evidence of exertional myopathy, even though dog muscle PFK activity is nearly absent, because dogs do not rely on anaerobic glycolysis for generation of energy during exercise. During exercise, PFK-deficient dogs do exhibit less extraction of oxygen from hemoglobin than normal dogs do, either because the affinity of hemoglobin for oxygen is high (caused by low 2,3-BPG levels in their erythrocytes) or because oxidative metabolism is impaired.
PFK activity in erythrocytes from newborn infants is about 50% to 60% of that in normal adult cells. PFK deficiency is more evident in older cells made earlier in gestation, perhaps because of accelerated enzyme decay. Vora and Piomelli showed that 25% to 30% of newborn erythrocyte PFK consists of L4 isozyme, with the remainder being divided equally between three hybrid isozymes of L and M subunits (LlM3, L2M2, and L3Ml). The L4 isozyme, not found in normal adult red cells, is unstable and presumably accounts for the reduced PFK activity of older cord red cells. The demonstration that PFK deficiency may result in hemolytic anemia in adults suggests that the enzyme deficiency characteristic of normal newborn red cells may contribute to their shortened survival.
Genetics and Inheritance
The gene locus for the L subunit of PFK has been assigned to chromosome 21, whereas the M subunit locus is on chromosome 12. The erythrocytes, but not the leukocytes and platelets of individuals with trisomy 21, consistently contain increased PFK activity. Increased erythrocyte PFK activity is caused by increased amounts of L subunit, consistent with a simple gene dosage effect.
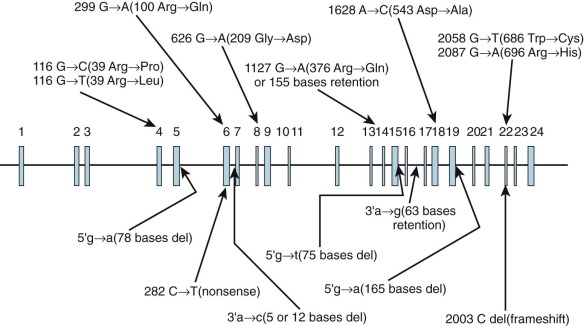
Inheritance is autosomal recessive. Type I PFK deficiency (Tarui disease), which is found predominantly in Ashkenazi Jews and Japanese individuals, is the result of mutations involving the PFKM gene. The 15 reported mutations (5 splice site, 8 missense, 1 nonsense, and 1 frameshift) are shown in Figure 17-3 . The widely scattered location of the mutations makes it difficult to correlate genotype and phenotype. In PFK-deficient dogs, a missense mutation involving 120 nucleotides from the 3′ end of the coding region produces a truncated mRNA and an unstable PFKM subunit tetramer with altered kinetic properties.
Therapy
Supportive care with red cell transfusions as needed and daily folic acid supplementation are the mainstays of therapy for hemolytic anemia.
Aldolase Deficiency
Clinical Manifestations, Biochemistry, and Genetics
Aldolase A is found in erythrocytes, muscle, and the fetal brain. Only six individuals with CNSHA and erythrocyte aldolase deficiency have been reported. Their clinical course has varied, with one also exhibiting mental retardation, three an associated myopathy, and two no abnormalities other than hemolytic anemia ( Table 17-5 ). Two members of a Japanese family who had a severe deficiency of red cell aldolase exhibited severe chronic hemolytic anemia, sometimes exacerbated by infections. In vitro, red cell glycolysis and HMP shunt activity were depressed, thus indicating that the deficiency of aldolase was of functional significance. Both parents were hematologically normal but had intermediate reductions in red cell aldolase A activity. The mutant aldolase was strikingly thermolabile. A missense mutation ( *GAT → *GGT ) giving rise to a single amino acid substitution (Asp→Gly) at position 128 was present in the mutant enzyme. Transfection of Escherichia coli with an expression plasmid containing normal, mutant, or modified (by site-directed mutagenesis) aldolase A–complementary DNA (cDNA) generated functional aldolase molecules that were used to confirm that the amino acid substitution at position 128, a site distant from the catalytic site but within an exposed hinge region, was responsible for enzyme thermolability.
| Ethnicity | Genotype | Amino Acid Change | Aldolase Activity ‡ | Thermolability | Anemia | Myopathy | Mental Retardation | Reference |
|---|---|---|---|---|---|---|---|---|
| Jewish | * | Unknown | 15 | NR | Yes | No | Yes | |
| Japanese | † | Unknown | 7 | Increased | Yes | No | No | |
| Japanese | Homozygote | Asp128→Gly | 5 | Increased | Yes | No | No | |
| German | Homozygote | Glu206→Lys | 4 | Increased | Yes | Yes | No | |
| Sicilian | Compound | Cys338→Tyr | 23 | Increased | Yes | Yes | No | |
| Italian | Heterozygote | Gly346→Ser | NR | Normal | No | Yes | No |
* Presumed homozygote because the parents were first cousins.
† Homozygote or compound heterozygote.
‡ Aldolase activity in erythrocytes expressed as percentage of normal.
In a German child, homozygosity for a point mutation at residue 206 (Glu→Lys) in the coding region of aldolase A also led to thermolability and severe enzyme deficiency in both erythrocytes and skeletal muscle. Unlike the Japanese subjects, this child had a metabolic myopathy, as well as mild to moderate hemolytic anemia. The mutation at residue 206 would be predicted to disrupt the main subunit interface of the aldolase tetramer and cause thermolability. Why myopathy was noted in one setting but not in the other is unknown.
A girl of Sicilian ancestry whose red cell aldolase activity was less than a quarter of normal also exhibited myopathy and chronic hemolytic anemia. Elevated plasma creatine phosphokinase activity was noted with febrile illnesses. She died at the age of 54 months of rhabdomyolysis and hyperkalemia during an acute febrile illness. Like the German patient, her aldolase A enzyme was thermolabile, which offered an explanation for the exacerbation of myopathy during fever. DNA analysis revealed two mutations in the aldolase A gene in this patient: one was a premature stop codon mutation ( 931*C → T ), and the second was a missense mutation ( 1037*G → A ) that conferred thermolability on the enzyme.
Another patient with aldolase deficiency exhibited CNSHA, mental retardation, mild glycogen storage disease, intestinal lactase deficiency, growth retardation, and peculiar facial features. Enzyme activity was approximately 15% of normal in erythrocytes and cultured skin fibroblasts. No structural abnormality of residual erythrocyte aldolase was detected by electrophoresis, isoelectric focusing, heat stability studies, or kinetic examination. The patient was the offspring of a consanguineous marriage, but both parents were hematologically normal and had normal red cell aldolase activity.
A sixth patient, heterozygous for a thermostable mutation of aldolase A, had myopathy, arthrogryposis multiplex congenita, and hypopituitarism but normal IQ and no evidence of hemolytic anemia. Because both parents were clinically normal, the aldolase mutation in the patient may have no relationship to his clinical abnormalities.
Triose Phosphate Isomerase Deficiency
Clinical Manifestations
An association with TPI deficiency has been documented in approximately 50 patients with congenital hemolytic anemia. In addition to chronic hemolysis, a severe neuromuscular disorder characterized initially by spasticity and psychomotor retardation and often progressing to weakness and hypotonia has been found in nearly all patients surviving beyond the neonatal period. Increased susceptibility to bacterial infection has also been noted. The neurologic abnormalities are not usually manifested before 6 to 12 months of age and are progressive, with death occurring before the age of 5 years. Occasionally, these abnormalities may stabilize during childhood or adolescence. In a Hungarian family, one adult son with severe TPI deficiency had extrapyramidal neurologic symptoms, whereas his older brother, who was equally TPI deficient, remained free of symptoms. Further investigation has shown that these brothers both inherited the same two TPI mutations (see “Genetics and Inheritance” section) but that levels of dihydroxyacetone phosphate (DHAP), methylglyoxal, and advanced glycation end products were much higher in the neurologically affected brother. The basis for the prolonged survival of both brothers and their different neurologic status is unknown, but epigenetic factors are suspected.
Anemia is variable, but most patients require at least occasional blood transfusions. Macrocytosis and polychromatophilia are evident on the blood smear because of the presence of reticulocytosis, which may reach 50% on occasion. Aside from occasional small, dense spiculated cells, no striking changes in erythrocyte morphologic characteristics are present.
Biochemistry
TPI is a homodimer whose two subunits (relative molecular mass: 26,750 ) are the product of a single locus on the short arm of chromosome 12. Posttranslational modification of one or both subunits may occur by deamidization of aspartines at positions 15 and 71, thereby resulting in multiple forms of the enzyme. The 248–amino acid sequence of human (placental) TPI has been determined directly and is nearly, but not completely, identical to the sequence predicted by nucleotide analysis of adult human liver cDNA.
TPI is a classic housekeeping gene, present in all tissues, with an amino acid sequence that has been remarkably well conserved during evolution. The eight-stranded αβ-barrel structure of the human enzyme has been confirmed at 2.8-nm resolution by x-ray crystallography. TPI has no requirement for cofactors or metal ions, and there is no evidence of cooperativity or allosteric interactions between subunits.
When measured in vitro, erythrocyte TPI activity is approximately 1000 times that of HK, the least active glycolytic enzyme. Even TPI-deficient erythrocytes exhibiting only 2% to 35% of normal TPI activity in vitro possess far more TPI than HK activity. Not surprisingly, TPI-deficient erythrocytes are capable of normal glycolysis in vitro, even when compared with reticulocyte-rich normal blood. Furthermore, mathematical modeling shows that increased activity of glycolytic kinases can lead to normal conversion of glucose to lactate in TPI-deficient cells. Nonetheless, DHAP, the substrate for TPI, accumulates to high levels in TPI-deficient erythrocytes, and the ATP concentration is usually low for cell age. These results indicate the presence of a substantial impairment in glycolysis in vivo. Reduction of residual TPI activity in deficient cells by binding to the red cell membrane or to brain microtubulin has been proposed to account for some of the differences between in vitro determinations of TPI activity and the evidence of a more severe impairment in enzyme function in vivo. The defect can be partially bypassed by way of the HMP shunt, which generates glyceraldehyde 3-phosphate from glucose without the participation of TPI. Methylene blue stimulation of the shunt produces a lesser increase in glycolysis relative to baseline in TPI-deficient erythrocytes than in reticulocyte-rich control blood. This has been interpreted as indicating a markedly greater “resting” shunt rate in the deficient cells, consistent with the proposed reliance of such cells on the shunt. Evidence indicates that TPI-deficient red cells are relatively deficient in antioxidant capabilities and that this deficiency may contribute to their shortened life span.
Rare electrophoretic variants of TPI, not associated with reduced enzyme activity or hemolysis, have been described. In anemic patients, enzyme kinetics and electrophoretic mobility are usually normal, but in vitro evidence of enzyme lability after heating is often obtained, thus suggesting that instability and rapid loss of enzyme protein play an important role in the pathogenesis of hemolysis in vivo. A single amino acid substitution (Glu→Asp) at position 104 has been identified in at least 17 geographically widely distributed families with clinically affected children and is the most commonly encountered mutation responsible for TPI deficiency. Its relative rate of occurrence is thought to be the result of a founder effect. The Glu104→Asp TPI gene product is thermolabile. Computer modeling indicates that a substituted amino acid at position 104, which is buried in a hydrophilic side pocket of the normal enzyme, will reduce the stability of the pocket, promote unfolding, and enhance thermolability. Comparison of the recombinant mutant TPI enzyme to the recombinant wild type indicates that enzyme kinetics are normal but that dimer formation and thermal stability are greatly decreased. Analysis of the crystal structure of the mutant TPI suggests that a mutation-triggered disruption of the water network in the vicinity of the dimer–dimer interaction is the underlying basis for enzyme instability and ultimately for the clinical abnormalities.
Fourteen TPI mutations have been defined. Most clinically affected individuals are homozygous for a single mutation or compound heterozygous for two different mutations. In the Hungarian pedigree mentioned earlier, a point mutation at codon 240 (Phe→Leu) produced a moderately thermolabile TPI with abnormal substrate kinetics and electrophoretic migration. Phe240 is near the active site of the enzyme and appears to be essential for maintaining its correct geometry. Furthermore, both anemic brothers in this pedigree inherited a nonsense mutation at codon 145 that terminated TPI protein synthesis, thereby producing a truncated protein and also reducing the output of TPI mRNA from the affected allele by 10- to 20-fold. The red cells of the brothers had less than 10% of the TPI activity found in normal cells.
The enzyme deficiency is manifested not only in red cells but also in leukocytes, platelets, muscle, serum, and cerebrospinal fluid. Histologic examination of muscle from a girl with TPI deficiency and myopathy revealed marked degenerative changes of the contractile system, altered mitochondria, and absent TPI by histochemical staining. Brain and nerve tissue have not yet been analyzed, but the cerebrospinal fluid deficiency suggests that deficient TPI activity in neural tissue is responsible for the neurologic abnormalities observed in enzymopenic patients. Orosz and coworkers have advanced the hypothesis that mutant TPI is aggregation prone and that such aggregates might be neurotoxic, but direct evidence for this is not available. Although increased susceptibility to infection might be the consequence of defective function by TPI-deficient leukocytes, functional studies carried out on such cells in vitro have often been normal. A functional defect in TPI-deficient platelets has been described.
Genetics and Inheritance
Several large pedigrees display an autosomal recessive mode of inheritance of TPI deficiency. Obligate heterozygotes are clinically normal, but their erythrocytes contain only approximately half the TPI activity of control erythrocytes. As is often the case in other glycolytic enzymopathies, no clear boundary exists between heterozygous deficient and low-normal enzyme activity. Several upstream polymorphisms are found at a high rate of occurrence in African-American populations. Although they may be associated with mild reductions in erythrocyte TPI activity, they do not seem to be associated with clinical abnormalities, even in homozygotes. At the other extreme, the homozygous state in mice for a TPI null allele is lethal early in embryogenesis. A more informative mouse model results in an Asp→Gly substitution at codon 49 located in the functional domain of TPI. The mutant enzyme is highly thermolabile, resembling in this regard the human Glu104→Asp TPI mutant. Homozygous mice have only 13% of normal TPI activity and exhibit hemolytic anemia. Simultaneous heterozygous inheritance of TPI deficiency and either G6PD deficiency or sickle cell trait has not altered the typical clinical pattern of the disorders when either is present alone. TPI deficiency can be diagnosed prenatally by analysis of fetal blood cells, cultured amniocytes, or trophoblastic cells with suitable precautions. If the mutation is known, mutation analysis can be performed directly on fetal DNA.
Therapy
Transfusions and folic acid supplementation are the therapies presently available. Splenectomy in one patient did not alter the intensity of hemolysis. In vitro studies point to the possibility of transfer of TPI enzyme protein from normal to deficient cells with transient improvement in enzyme activity and a reduction in DHAP levels. There have been no attempts to deliver normal TPI enzyme to patient tissues, but in other disease settings direct enzyme transfer from normal hematopoietic stem cell–derived microglial cells to enzyme-deficient nervous tissue after allogeneic stem cell transplantation has been successful.
Glyceraldehyde-3-Phosphate Dehydrogenase Deficiency
Clinical Manifestations, Biochemistry, and Genetics
Study of a large kindred in which three members exhibited a reduction in erythrocyte G3PD activity to 50% of normal levels yet were hematologically normal have clearly established that G3PD deficiency need not result in hemolysis. The clinical severity of hemolytic anemia in four members of this pedigree who inherited both hereditary spherocytosis and G3PD deficiency was no greater than that associated with spherocytosis alone. Affected members of this kindred were presumed to be heterozygotes, because the amounts of both G3PD enzyme activity and enzyme protein were reduced equally to about 50% of normal and the amount of residual G3PD was qualitatively normal. Two of three patients who had anemia with G3PD deficiency in other kindreds had even lower levels of enzyme activity (20% to 30% of normal) and conceivably were either homozygotes or doubly heterozygous for two mutant G3PD genes. However, as with the spherocytosis pedigree, a cause-and-effect relationship between hemolytic anemia and G3PD deficiency was not established.
Phosphoglycerate Kinase Deficiency
Clinical Manifestations
PGK deficiency is a sex-linked disorder that largely affects males. Nonspherocytic hemolytic anemia may occur alone or in combination with neurologic abnormalities ranging from emotional instability to seizures, movement disorders, psychomotor retardation, aphasia, or tetraplegia. In a subset of individuals with PGK deficiency, myopathy is the dominant and sometimes the only clinical manifestation ( Table 17-6 ). Individuals with one form of mild PGK deficiency (PGK München) exhibit no clinical abnormalities (see Table 17-6 ). In females, erythrocyte PGK activity is less depressed than it is in males, hemolytic anemia is mild or absent, and neurologic and muscle abnormalities are not seen (see Table 17-6 ).
| Variant | Mutation * | RBC PGK Activity (% of Normal) | Stability In Vitro | Kinetic Abnormalities | Hemolytic Anemia | Neurologic Abnormalities | Myopathy |
|---|---|---|---|---|---|---|---|
| Hemizygotes (Male): Hemolytic Anemia or No Clinical Manifestations | |||||||
| New York | Asp164→Val | 2-2.7 | Normal | + | + | 0 | |
| Alabama | Lys190 or 91 del | 4 | + | 0 | 0 | ||
| Matsue | Leu89→Pro | 5 | Low | + | + | + | 0 |
| Uppsal | Arg206→Pro | 5-10 | Low | + | + | + | 0 |
| Cincinnati | 8-11 | + | + | 0 | |||
| Tokyo | Val266→Met | 10 | Low | + | + | + | 0 |
| Michigan | Cys315→Arg | 10 | Low | + | + | + | 0 |
| Barcelona | Ile147→Asn | 10 | + | + | 0 | ||
| San Francisco | 12 | Normal | + | + | 0 | 0 | |
| München | Asp267→Asn | 21 | Low | 0 | 0 | 0 | 0 |
| Murcia | Ser319→Asn | 49.0 | + | + | 0 | ||
| Herlev | Asp285→Val | 50 | + | 0 | 0 | ||
| Hemizygotes (Male): Muscle Disease | |||||||
| Shizuoka | Gly158→Asp | 0.7 | Normal | 0 | + | 0 | + |
| North Carolina | 10–amino acid insert | 3 | Normal | + | 0 | + | + |
| Creteil | Asp315→Asn | 3 | Low | + | 0 | 0 | + |
| Trondheim | 5 | 0 | 0 | + | |||
| Antwerp | Glu252→Ala † | 5.6 | 0 | 0 | + | ||
| Fukui 246 | 4-bp del, exon 6 § | 5.6 | 0 | 0 | + | ||
| +Kyoto | Ala354→Pro | 6 | + | +/− | + | ||
| Hammamatsu | 253Ile→Thr | 8 | Normal | 0 | 0 | + | + |
| New Jersey | 18 | Normal | + | 0 | 0 | + | |
| Alberta | 1.5 ‡ | Normal | + | 0 | 0 | + | |
| PGK Afula2 | Thr378→Pro | 2 | 0 | 0 | + | ||
| Heterozygotes (Female) | |||||||
| Piedmont | 27 | + | 0 | 0 | |||
| Memphis | 78 | + | 0 | 0 | |||
| Amiens | 77 | + | 0 | 0 | |||
* Mutation position as corrected by Beutler.
† This mutation also adversely affects mRNA splicing efficiency to about 10% of normal.
§ Frameshift at codon 706-709 resulting in a stop codon truncating phosphoglycerate kinase to 231 amino acids in length.
Hematologic findings in individuals who have anemia with PGK deficiency have been those customarily associated with hemolysis, namely, jaundice and reticulocytosis. Hemolytic episodes are often associated with acute febrile illnesses. No changes in erythrocyte morphologic characteristics have been seen, and osmotic fragility has usually been normal.
Biochemistry
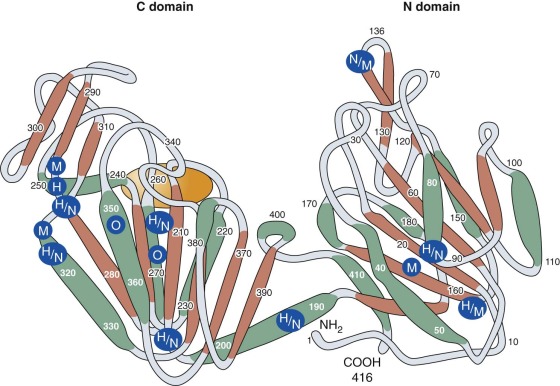
PGK is a monomeric enzyme with a primary structure consisting of 417 amino acids. Horse and pig muscle PGK has a primary structure that is highly homologous to that of human PGK. Crystallographic studies show that its tertiary structure consists of two lobes (the C and N domains) connected by a hingelike structure that allows considerable conformational change to occur during substrate binding ( Fig. 17-4 ). Because the nucleotide (adenosine diphosphate [ADP] and ATP) combining site is located within the C domain and the phosphoglycerate (1,3-BPG or 3-phosphoglyerate) binding site is within the N domain, bending of the enzyme is required to bring the several substrates required for the PGK reaction into close proximity. Mutant PGKs often exhibit abnormal enzyme kinetics and diminished stability in vitro, properties that are likely to impair enzyme function in vivo (see Table 17-6 ).
PGK-deficient red cells are capable of normal glycolysis in vitro. Intracellular ATP concentrations are normal or slightly low, whereas the 2,3-BPG concentration is elevated, sometimes to two or three times the normal level. These results reflect increased flow through the 2,3-BPG cycle (see Fig. 17-1 ) at the expense of the ATP-generating PGK reaction. Despite the availability of an alternative pathway (the Rapoport-Luebering shunt or the 2,3-BPG cycle) to bypass PGK, substantial accumulation of glycolytic intermediates proximal to the enzyme defect is found in fresh red cells, thus indicating that the normal flow of erythrocyte glycolysis is impeded in vivo.
Little is known about the mechanism of hemolysis of PGK-deficient red cells. Most PGK activity is membrane associated. It has been suggested that the ATP for membrane adenosine triphosphatase (ATPase)–mediated cation transport is mostly (or entirely) generated by membrane-bound PGK. Indeed, ADP derived from membrane ATPase exerts an important regulatory influence on glycolysis by its participation in the PGK reaction. However, the implication of a special role for PGK in cation transport has been challenged. Active transport of Na + and K + by PGK-deficient red cells with residual PGK activity only 10% to 15% of normal was not impaired, even with the challenge of an increase in intracellular Na + concentration. Thus it seems unlikely that hemolysis of PGK-deficient red cells is related to premature cation pump failure as a direct result of inadequate PGK activity.
PGK activity in leukocytes is consistently subnormal in affected males, but white cell function is not usually compromised.
Genetics and Inheritance
The major structural gene for PGK, located on the long arm of the X chromosome, is 23 kb in size and composed of 11 exons and 10 introns. PGK-deficient male hemizygotes demonstrate very little active enzyme and have more symptoms than heterozygous females do, who have higher levels of PGK activity (see Table 17-6 ). A second functional PGK gene, expressed only in spermatozoa, is found on chromosome 19. This autosomal PGK gene lacks introns but is otherwise similar to the X-chromosomal PGK gene. A single isozyme of PGK is found in all human nonhematopoietic tissues except spermatozoa. It is therefore not surprising that nonerythroid tissues may be compromised in patients with erythrocyte PGK deficiency.
In 1972, Yoshida and coworkers succeeded in purifying and sequencing both normal erythrocyte PGK and a clinically normal but electrophoretically distinct human mutant PGK, the New Guinea variant. The mutant enzyme differed from normal PGK by the substitution of arginine for threonine at position 352. Subsequently, the structure of 17 other PGK mutants, most associated with clinical manifestations, has been determined by peptide or nucleotide sequencing (see Table 17-6 ). Fifteen of these mutants involve only a single amino acid (14 missense mutations and 1 deletion). The 16th activates a cryptic splice site that leads to the insertion of 10 additional amino acids into the PGK polypeptide, and the 17th, a 4-amino acid deletion, also affects splicing and produces a truncated polypeptide. The majority of the mutants are found within the C domain, as shown in Figure 17-4 . It is not clear how the particular spectrum of clinical abnormalities associated with each mutation is related to its position within the molecule. For example, PGK Michigan, a point mutation at amino acid 316, is associated with hemolytic anemia and neurologic abnormalities, whereas individuals with PGK Creteil, a point mutation of the adjacent amino acid at position 315, do not have anemia and neurologic abnormalities but exhibit rhabdomyolysis. Both mutants are thermolabile and demonstrate kinetic abnormalities in vitro, and both have similarly reduced activity in erythrocytes (see Table 17-6 ). Random inactivation of the mutant X chromosome may produce differing proportions of enzyme-deficient cells in female heterozygotes. Some may be anemic (see Table 17-6 ), whereas others are clinically and hematologically normal. In the latter, the population of PGK-deficient red cells may be so small that erythrocyte PGK activity will be completely normal.
Therapy
In one patient, splenectomy had no beneficial effect on anemia, but in several others, surgery has decreased or eliminated the need for transfusions, reduced the degree of reticulocytosis, and sometimes resulted in an increase of several grams in the hemoglobin level.
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


