Fig. 7.1
Generation of extracellular ATP and adenosine in the tumor microenvironment (TME). (1) In response to hypoxia, cells in the TME release ATP through either ATP-binding casettes, Pannexin-1 or connexins. (2) In addition, the purinergic receptor, P2X7R can also contribute towards the secretion of ATP in the TME. (3) Accumulation of ATP in the milieu stimulates the P2 purinergic receptors; P2XR and P2YR. (4) The sequential action of CD39 and CD73 degrade ATP to ADP/AMP and adenosine (Ado) respectively, thus contributing majorly to the generation of an immunosuppressive TME. (3) Some P2YR including, P2Y1R, P2Y12R and P2Y13R are able to bind ADP and trigger downstream signaling. Ado modulate tumor growth by acting through P1 receptors, A1R, A2AR, A2BR and A3R. (5) The accumulated adenosine can be subsequently catabolized to inosine in the presence of adenosine deaminase (ADA) and its receptor CD26. ATP adenosine triphosphate, ADP adenosine diphosphate, AMP adenosine monophosphate
During neoplasia, extracellular adenosine is generated predominantly via the sequential activation of two Ecto-nucleotidases. These include CD39, which hydrolyzes ATP and ADP to AMP, and CD73 that dephosphorylates AMP further to adenosine (Fig. 7.1). Transcription factors Sp-1 and HIF-1α mediate the upregulation of CD39 and CD73 expression, [23]. Besides CD39 and CD73, alkaline phosphatases (ALP) and several members of the nucleotide pyrophosphatase and phosphodiester family including NPP1/CD203a can also contribute to the production of extracellular adenosine. However, the involvement of this pathway in cancers is poorly understood, although it has been reported to be activated in gliomas, melanoma, prostate cancer [24], and multiple myeloma [25]. Importantly, adenosine can be further degraded to inosine in the presence of adenosine deaminase (ADA) through its association with CD26 (Fig. 7.1). A study by Tan et al., however, showed that elevated levels of adenosine itself could potently inhibit the activities of ADA and CD26 on HT-29 colorectal cancer cells [26]. It remains to be determined whether ADA is critical during cancer development.
7.3 Purinergic Receptors in Immunity and Cancer
The biological actions of ATP and adenosine rely on the activation of purinergic receptors that are expressed abundantly on both tumors and the infiltrating immune and non-immune cells. ATP binds to P2Rs while adenosine binds P1Rs. The P2Rs can be further classified into seven P2X (P2X1-P2X7) and eight P2Y (P2Y1–2, P2Y4, P2Y6, P2Y11–14) receptor subtypes. P1Rs bind adenosine and comprise of four G-protein coupled receptors (GPCRs), namely A1R, A2AR, A2BR, and A3R [10]. Importantly, the concerted action of the two Ecto-nucleotidases, CD39 and CD73 constitute the major source of extracellular adenosine in the TME. In this section, therefore, we discuss how activation of the purinergic receptors as well as Ecto-nucleotidases on tumor cells and immune cells influences cancer growth and metastasis.
7.3.1 Effect on Tumor Cells
7.3.1.1 ATP Sensing P2 Receptors: P2XR and P2YRs
The high ATP concentration in the TME can directly affect the expression and function of P2Rs during tumor development. This is evident by the differential expression of P2Rs on several human and mouse cancer cell lines (summarized in Table 7.1). Typically, the expression of P2XRs on tumor cells is associated with anti-tumor functions (see Table 7.1). However, P2X7R has seemingly opposite effects on the growth and survival of tumor cells. For example, activation of P2X7R facilitated the migration of PC9 lung carcinoma and T47D breast cancer cells and siRNA knockdown of P2X7R in these cells reversed their migration ability [38]. Conversely, exogenous addition of ATP to P2X7R overexpressing acute myeloid leukemia (AML) cells repressed its expression and resulted in the inhibition of cell-cycle genes, migration, and adhesion molecules [71]. Likewise, activation of P2X7R on B16F10 melanoma cells resulted in reduced proliferation and survival of these cells [72]. A possible explanation for the disparities in these findings could be attributed to the levels of ATP used for in vitro stimulation in these studies. This contention was supported by Giannuzzo et al., who found the concentration of ATP could have opposite effects on P2X7R activity. Specifically, high levels of ATP activated P2X7R on human pancreatic ductal adenocarcinoma cells (PDAC) resulting in their reduced proliferation and survival. By contrast, low or basal levels of ATP promoted their cell migratory and invasive properties [37]. Such opposing P2X7R effects were also noted in cancer patients, whereby expression of P2X7R correlated favorably to overall survival in patients with gliomas, non-small cell lung cancer (NSCLC), and hepatocellular carcinoma (HCC). The reverse was, however, seen in patients with renal cell carcinoma (RCC) (Table 7.2 ).
Table 7.1
Expression of purinergic receptors on cancer cells
Type of purinergic receptor | Receptor | Human cancer cells | Mouse cancer cells | Functions | References |
|---|---|---|---|---|---|
P2XR | P2X3 | Colon carcinoma (SW480) | – Inhibition of apoptosis | [27] | |
P2X4 | Mammary carcinoma (MDA-MB-231), non-small cell lung cancer (NSCLC) patient tumors | Mammary carcinoma (4T1.2) | – Activation of autophagy and ICD | ||
P2X5 | Lymphoid malignancies, melanoma (A431) | – P2X5R+ lymphoid tumors are easily lysed by Lrh-1+ CTLs – Inhibition of cell proliferation | [30] | ||
P2X7 | Mammary tumors (MDA-MB-231, MCF-7, MCF-10A, T47D) NSCLC patient tumors Pancreatic ductal adenocarcinoma (PDAC) Mesothelioma Prostate cancer Lung cancer cells (pc-9, H292) Pancreatic cancer (Panc-1, MIA PaCa-1) Ovarian cancer (SK-OV3, Caov-3, MM14.Ov) | Mammary carcinoma (4T1.2) Melanoma (B16F10) Colon carcinoma (MCA38) | – Activation of autophagy and ICD – Increased cell migration and invasion – Promotes proliferation – Promotes epithelial-mesenchymal transition (EMT) – Promotes cell death – Increases Ca+2 levels | ||
P2YR | P2Y1 | Prostate cancer (PC-3) | – Increase in cell number – Increase migration through ERK pathway | ||
P2Y2 | Melanoma (A431), Mammary tumors (MDA-Mb-231, MCF-7) Prostate cancer (pc-3 M) Ovarian cancer (SK-OV3) Lung epithelial carcinoma (A549) | – Increase in cell number – Increase migration through ERK pathway – Increased IL-8 and invasion – Activation of EGFR and induction of EMT – Increase expression of adhesion molecules | |||
P2Y6 | Mammary carcinoma (MDA-MB-231) | – Invasion and cancer migration | [39] | ||
Ecto-nucleotidase | CD39 | B cell lymphoma (BL-41, B104, Ramos) Melanoma (SK-Mel-5, SK-Mel-28) B CLL (EHEB, MEC2) Ovarian cancer (OAW-42, Sk-OV3) | – Increased ATPase activity – Inhibition of CD4+T cell and CD8+T cell proliferation – Inhibition of NK cell cytotoxicity | ||
CD73 | Endometrial cancer (G1 EEC, HEC-1A, HEC-1B) Breast cancers (MDA-Mb-231, LM3) | Melanoma (SM1WT1, B16F10a) Mammary tumors (4T1.2) | – Increased expression on tumor core in A2AR-deficient mice – In endometrial cancers – CD73 is essential to prevent tumor cell migration and invasion – In B16F10 cells, CD73 inhibited cell migration and adhesion – Promotes extravasation and colonization in breast cancer cells | ||
Adenosine receptor | A1R | Endometrial cancer (G1 EEC, HEC-1A, HEC-1B) Glioblastoma patients Colon cancer (CW2) Breast cancer (MCF-7) | Glioblastoma (Gl261) | – In endometrial cancers, CD73 is essential to prevent tumor cell migration and invasion via A1R – Reduced tumor cell proliferation and induction of glioma cell death – Regulates expression of estrogen receptor-α on MCF-7 cells and induces tumor proliferation | |
A2AR | Ovarian cancer (OVCAR-3, Caov-4, SCOV-3) NSCLC cell lines Melanoma (A375) | – cAMP induction – Induction of tumor cell proliferation | |||
A2BR | Mammary cancer (MDA-MB-231) Gastric cancer (HGC-27) Ovarian cancer (OVCAR-3, Caov-4) Prostate cancer (PC-3, DU145, LnCaP) | Melanoma (B16F10) Mammary tumor (4T1.2, E0771) | – Induces FGF-2 and B16F10 cell proliferation – A2BR is a target of Fos-related antigen-1 (Fra-1) and mediates metastasis – Promotes proliferation, migration, and invasion | ||
A3R | Ovarian cancer (OVCAR-3, Caov-4) Melanoma (A375) Colon cancer (HCT 116, HT-29) Renal cell carcinoma (RCC4-VHL, ACHN) Glioma cells (A172) Bladder cancer cells (253 J and 5637, T24) Breast cancer (MCF-7, MDA-Mb-231) Prostate cancer (DU-145, PC3, LNCap-FGC-10) | Colon cancer (CT-26) Melanoma (B16-BL6) Lewis lung carcinoma (LLC) Leukemia (HL60) | – Inhibition of tumor growth – Promote tumor cell apoptosis via caspase-3 and caspase-9 – Majorly inhibits but is shown to promote growth of HT29 cells – Inhibition of renal cell carcinoma growth – Increased tumor death via increased ROS production and intracellular Ca+2 – Reduced tumor cell migration |
Table 7.2
Role of purinergic receptors in human cancers
Type of purinergic receptor | Receptor | Cancer type | Major finding | References |
|---|---|---|---|---|
ATP receptors | P2X3 | Hepatocellular carcinoma (HCC) | – Low expression of P2X3R was associated with increased OS and RFS in these patients (n = 188) | [73] |
P2X7 | Non small cell lung cancer (NSCLC) | – No association between P2X7R expression and survival in these patients (n = 93) | [74] | |
P2X7 | NSCLC | – High expression was associated with better RFS and OS (n = 96) | [75] | |
P2X7 | Glioma | – High expression of P2X7 was associated with longer survival and increased sensitivity to radiotherapy (n = 343) | [76] | |
P2X7 | HCC | – Increased peritumor P2X7 was associated with favorable prognosis in patients after surgical resection (n = 273) | [77] | |
P2X7 | Renal cell carcinoma | – Low intratumoral P2X7R was associated with better RFS in patients (n = 273) | [78] | |
P2Y13 | HCC | – High expression was associated with increased OS and RFS (n = 188) | [73] | |
Ecto-nucleotidase | CD39 | High-grade serous (HGS) ovarian cancer | – High CD39 expression showed a trend (p = 0.0507) towards poor OS (meta-analysis) | [79] |
CD39 | HCC | – High CD39 expression in HCC tumors from 326 patients correlated with poor OS and RFS | [80] | |
CD39 | Gastric cancer | – Overexpression of CD39 in tumor but not peritumor was associated with poor prognosis in these patients – Similarly high CD39 expression on CD8+T cells was associated with poor survival outcomes (n = 84) | [81] | |
CD39 | Chronic lymphocytic leukemia (CLLL) | – Patients with high CD39 expression on T cells but not B cells showed poor survival (n = 68) | [82] | |
CD73 | Breast cancer | – High CD73 expression was correlated with poor OS and DFS (n = 136) | [83] | |
CD73 | Gastric cancer | – High CD73 expression was associated with poor prognosis in these patients (n = 68) | [84] | |
CD73 | Triple negative breast cancer (TNBC) | – High CD73 expression was associated with poor prognosis in TNBC patients but no association was identified in patients with HER2+ tumors or luminal breast cancers | [85] | |
CD73 | NSCLC | – High CD73 expression was associated with poor prognosis (n = 653) | [86] | |
CD73 | Human rectal adenocarcinoma | – High tumoral CD73 was associated with worse outcomes (n = 90) | [87] | |
CD73 | Ovarian cancer | – High CD73 expression was a poor prognostic marker in these patients – This correlated with increased infiltration of CD73+CD8+T cells in the tumor (meta-analysis) | [79] | |
CD73 | Prostate cancer | – High expression was associated with shorter RFS and shorter bone-metastasis free survival in these patients (n = 285) – These observations correlated with increased CD73 expression on tumor infiltrating CD8+T cells | [88] | |
CD73 | Oral squamous cell carcinoma (OSCC) | – High CD73 expression was associated with poor RFS and overall survival in patients with OSCC (n = 113) | [89] | |
CD73 | Head and neck squamous carcinoma (HNSC) | – High CD73 impacted OS in these patients (n = 162) and was associated with high lymph node metastasis | [90] | |
CD73 | Urothelial bladder cancer | – High CD73 expression reduced the rate of progression of cancer in these patients (n = 174) | [91] | |
Adenosine receptor | A2AR | NSCLC | – Better OS and RFS was observed in patients with high A2AR expression (n = 653) | [86] |
A2BR | TNBC | – High A2BR expression was associated with poor survival in TNBC patients but not in HER2+ or luminal breast cancer patients (meta-analysis) | [59] |
Expression of P2YRs on tumors largely favors tumor growth, migration, and survival (Table 7.1 ). P2Y2Rs were detected on the human breast cancer cell line MCF-7 and its activation potentiated migration and proliferative capability [40]. Similarly, the activation of P2Y2R on MDA-MB-231 cells increased the expression of adhesion molecules intercellular adhesion molecule-1(ICAM-1) and vascular cell adhesion molecule (VCAM). This subsequently resulted in increased adhesion of tumor cells to endothelial cells, thereby facilitating tumor cell migration and invasion [92]. By contrast, the migration of AML cells in response to the chemokine CXCL12 was blunted following incubation with P2Y2R and P2Y6R agonists [71].
7.3.1.2 Adenosine Generating Ecto-nucleotidases: CD39 and CD73
Co- or differential expression of CD39 and CD73 has been reported on many human cancers of solid and hematological origins, as summarized in Table 7.1. Accumulating evidence now suggests that the expression of CD39 and CD73 on tumor cells can potently trigger tumor cell growth and survival. It is hence not surprising that cancer patients with high expression of CD39 and/or CD73 show poor survival and recurrence-free survival outcomes across several cancer types (Table 7.2 ). In pre-clinical experiments, co-culture of human CD39+CD73+ SK-MEL5 melanoma cells with human peripheral blood mononuclear cells (PBMCs) impaired the proliferation of CD4+ and CD8+ T cells, as well as hampered the cytolytic activity of CD8+ T cells. Pre-treatment of cancer cells with a CD39 inhibitor rescued this suppressive phenotype and increased the effector functions of CD8+ T cells and NK cells [42]. Similarly, CD39 expression on human ovarian cancer cells was reported to suppress T and NK cell effector functions [93]. CD73 expressing ID8 mouse ovarian cancer cells also inhibited the proliferation of CD8+ and CD4+ T cells and the production of the effector cytokine IFN-γ [94]. Exosomes isolated from mesothelioma patients were also found to express CD39 and CD73, which exhibited potent enzymatic activity leading to production of local adenosine. Functionally, these exosomes suppressed human CD4+ and CD8+ T cell proliferation and cytokine release [95]. While CD73 is well characterized for its pro-tumor role, interestingly, a group recently identified an anti-tumor role for CD73 on human endometrial carcinoma. In this study, loss of CD73 on these tumors impaired actin polymerization, thus hampering cell-cell adhesion and loss of endometrial epithelial cell integrity. As a result, these tumors became highly migratory and invasive [48].
7.3.1.3 Adenosine Binding P1 Receptors: A1, A2a, A2b, and A3
P1Rs, similar to the P2YRs are widely expressed by tumor cells in mouse and humans (Table 7.1). Under physiological conditions, adenosine activates the high affinity A1, A2A, and A3 receptors. In contrast, activation of A2BR, a low affinity receptor occurs in conditions when adenosine concentration is high, which is frequently seen under pathological conditions such as the TME. Signaling via A1R and A3R inhibits intracellular cyclic AMP (cAMP), while A2AR and A2BR activate adenylyl cyclase and protein kinase A, leading to increased levels of (cAMP). High levels of cAMP is generally associated with profound immunosuppression and therefore A2AR and A2BR are viewed as tumor promoting adenosinergic receptors [96].
The suppressive function of A2AR has largely been investigated in the context of immune cells within the TME, and there exists limited understanding on how A2AR expression on tumors might influence tumor cell functions. A recent study reported that high expression of A2AR tumor biopsies of in adenocarcinoma patients correlated favorably with survival [86]. Paradoxical to this observation, inhibition of A2AR in human A375 melanoma cells reduced cell survival and proliferation in these tumors [10, 86]. Hence, the precise reason underlying these opposing results is presently unknown. A2BR, on the other hand, largely exerts its suppressive functions on tumor rather than immune cells. This receptor is detected in human and mouse triple negative breast cancer (TNBC) cells and its expression increased survival of these cells. In line with these findings, overexpression of A2BR was associated with poor survival in patients with TNBC, multiple myeloma, AML, and liposarcoma [59]. Wang et al. recently showed that microRNA-128b repressed the expression of A2BR. In patients with gastric cancer, microRNA-128b is downregulated leading to increased expression of A2BR, and this consequently increased tumor cell proliferation, migration, and survival [58].
Activation of A1R increases estrogen receptor-α (ER-α) expression on breast cancer cells and subsequently supports cancer growth [50]. The expression of A3R on tumor cells has contrasting effects. A3R is observed in primary human glioblastoma and prostate cancer, and in vitro stimulation inhibits proliferation of PC3 prostate carcinoma, HCT-116 colon carcinoma, and MIA-PaCa pancreatic carcinoma cells [97]. Paradoxically, activation of A3R on HT29 and CaCo2 colon cancer cells induces tumor cell proliferation [98]. Similarly, treatment with an A3R agonist CI-IB-MECA on human A375 melanoma cells induced the secretion of an angiogenic factor, Angiopoeitin-2 (Ang-2) [99].
7.3.2 Effects on Immune Cells
7.3.2.1 ATP/ADP Binding P2 Receptors
P2XRs mediate the transport of Na+, K+, and Ca2+ ions across the plasma membrane [10]. As a result, it was assumed that these receptors were not involved in the regulation of anti-tumor immunity. Therefore, although P2XRs and P2YRs were identified around the same time, the role of P2XRs in immune regulation is only now becoming apparent [10]. The expression of individual P2XRs on various immune cell types is outlined in Table 7.3. Of the known P2XRs, the expression and function of P2X7R has been most comprehensively investigated, in both immune and tumor cells. Cockcroft and Gomperts were the first to identify P2X7R expression in mast cells from rats [142]. They initially termed P2X7R as “ATP4− receptor,” because of its association to fully dissociated ATP (ATP4−) [142]. In 1996, this “ATP4− receptor” was cloned by Surprenant et al. and subsequently re-named as P2X7R, owing to its homology to other P2XRs [143]. P2YRs, by contrast, are G-protein coupled receptors that respond to ATP/ADP or even uridine 5′-phosphate (UTP) and influence the activation of (cAMP), which can either have pro- or anti-inflammatory functions [10]. Similar to P2XRs, P2YRs are also expressed in mouse and human immune cells, with the P2Y6R subtype being investigated more thoroughly than the others [10] (Table 7.3).
Table 7.3
Expression of purinergic receptors on human and mouse immune cells
Type of purinergic receptor | Receptor | Human immune cell subsets | Mouse immune cell subsets | Function(s) | References |
|---|---|---|---|---|---|
P2XR | P2X1R | B cells neutrophils, DC | ND | – Chemotaxis – DC maturation | |
P2X4R | B cells, T cells, Dc, Monocytes Macrophages | Macrophages | – T cell activation – DC maturation – Calcium mobilization – Induction of autophagy | ||
P2X5R | T cells B cells | ND | – T cell activation | [107] | |
P2X7R | B cells CD4+ T cells CD8+T cells Macrophages DC Tregs | Macrophages CD4+ T cells CD8+ T cells Tregs B cells | – CD62L shedding on T cells – Inflammasome activation – Tregs proliferation – T cell homeostasis – Shedding of CD23 on B cells | ||
P2YR | P2Y2R | Neutrophils Macrophages T cells | Macrophages Neutrophils DC Inflammatory monocytes | – Release of granules, activation of neutrophils – Neutrophil migration, release of inflammatory mediators – T cell activation – ROS production | |
P2Y6R | Monocytes, Macrophages, Neutrophils | Eosinophils | – Neutrophil migration – Release of pro-inflammatory cytokines – Increases responsiveness of leucocytes to CCL2 – Induce chronic allergic inflammation | ||
P2Y11R | Neutrophils NK cells DC | Only present in humans | – Activation marker – NK cell cytotoxicity and chemotaxis – Inhibition of IL-12 | ||
P2Y14R | Neutrophils | T cells Neutrophils | – Chemotaxis – Inhibition of T cell proliferation | ||
Ecto-nucleotidase | CD39 | Neutrophils Tregs B cells Tr-1 cells CD8+ T cells | B cells Tregs MDSC NKT cells T cells | – Inhibition of IL-8 – Induction of local immunosuppression – T cell exhaustion – Survival and cytokine release by NKT cells | |
CD73 | Tregs B cells Tr-1 cells Monocytes NK cells | B cells Tregs CD8+ T cells MDSC T cells NK cells | – Induction of local immunosuppression – Polarization to M2 macrophages – Inhibition of T cell cytokines – Inhibition of NK cell cytotoxicity | ||
Adenosine receptor | A1R | Monocytes neutrophils | – Secretion of VEGF by monocytes – Chemotaxis | ||
A2AR | Monocytes neutrophils | Macrophages CD4+ T cells CD8+ T cells NK cells | – M2-macrophage polarization – Inhibition of T effector functions – Chemotaxis – Inhibition of NK cell cytotoxicity | ||
A2BR | Monocytes DC CD4+ T cells CD8+ T cells | MDSC DC | – M2-macrophage polarization – TH2 polarization – Deactivation of T cells | ||
A3R | B cells neutrophils | – Inhibit B cell proliferation – Reduce neutrophil chemotaxis |
Similar to the effects on tumors, the activities of P2 receptors are largely modulated by the levels of extracellular ATP (Table 7.3) (Fig. 7.2). For example, low to moderate levels of ATP triggered mouse dendritic cell (DC) maturation and induction of T helper 2 (TH2) cells via the activation of P2Y11R [144]. High levels of ATP, by contrast, activated P2X7R on human alveolar macrophages and this interaction resulted in the release of the inflammatory cytokine IL-1β [115]. Vishva Dixit’s group later showed that activation of the inflammasome was responsible for the P2X7R-mediated IL-1β secretion [145]. Signaling via P2X7R can also activate several key macrophage function including production of reactive oxygen species (ROS), nitric oxide (NO), induction of phagocytosis, and formation of multinucleated giant cell formation [146]. Similar induction of migratory and chemotactic functions has been reported for P2Y2R, P2Y11R, and P2Y14R signaling on human and mouse neutrophils [147], and P2Y2R and P2Y6R on human monocytes, mouse macrophages, and DC [19]. On human NK cells, engagement of P2Y11R suppresses NK cell responsiveness to the chemokine CX3CL1, by abolishing their migratory and cytotoxic functions [124]. T cell receptor (TCR) stimulation can also trigger ATP release from T cells, which then mediates autocrine activation of P2X1R, P2X7R, and P2X4R to augment T cell proliferation and signaling [148]. Importantly, signaling via the P2YRs on human endothelial cells promotes angiogenesis through transactivation of vascular endothelial growth factor receptor-2 (VEGFR-2) [149, 150].
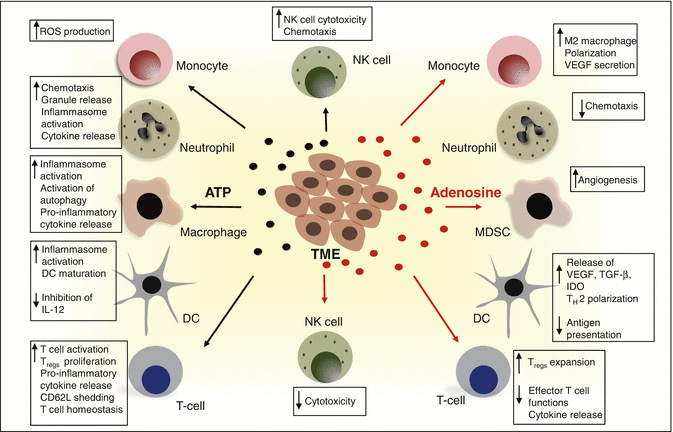
Fig. 7.2
The ecto-nucleotidase activities of CD39 and CD73 shifts the tumor dynamics from an ATP-driven inflammatory niche to an adenosine-rich immunosuppressive TME. ATP released from tumors and tumor infiltrating cells activates the P2 purinergic receptors. This primarily induces anti-tumor effects through the activation of the inflammasome, autophagy, increased cytokine release and activation of the immunogenic cell death (as shown in left). In contrast, the generation of adenosine through the CD39-CD73 axis negatively influences the TME and promotes tumor growth and metastasis via mechanisms such as increased angiogenesis, expansion of immunosuppressive immune populations (MDSC and Treg) and impaired cytotoxic functions of NK cells and CD8+ T cells (shown in right). ATP adenosine triphosphate, DC dendritic cell, NK natural killer, ROS reactive oxgen species, MDSC myeloid-derived suppressor cells, VEGF vascular endothelial growth factor, TGF-b transforming growth factor, IDO indoleamine 2,3 dioxygenase, Treg regulatory T cell
7.3.2.2 Adenosine Generating Ecto-nucleotidases: CD39 and CD73
Gregory et al. were the first to describe CD39 expression on B cells. In their study, CD39 was identified as an activation marker on tonsillar B cells from patients with Burkitt’s Lymphoma [151]. Since then, expression of CD39 has been found to be constitutively expressed in spleen, thymus, lung, as well as on primary tumors (Table 7.3). Cytokines, oxidative stress, and hypoxia-induced transcription factors such as Sp-1, STAT3, and zinc finger protein growth factor independent-1 transcription factors (Gfi1) can all upregulate CD39 expression [24]. The expression pattern of the other ecto-nucleotidase, CD73 on immune cells and tissues also closely resembles that of CD39 [9, 24] (Table 7.3).
The immunosuppressive effects of CD39 and CD73 can be attributed to the relatively high expression of these receptors on anti-inflammatory cells including tumor-associated macrophages (TAM), Tregs, type-1 regulatory (Tr-1) cells, and suppressive myeloid populations (Table 7.3 ). In the TME, the major functions of CD39 and CD73 lie in triggering the rapid catabolism of ATP to immunosuppressive adenosine to prevent the desensitization of P2 receptors and promote tumor development. Indeed, when co-incubated with CD4+ T cells, CD73+CD39+ TAM significantly suppressed the proliferation of T cells through production of adenosine. Blocking the activity of either CD39 or CD73 on TAM was sufficient to rescue this T cell effect [152]. Similarly, co-culture of CD39+ Tr-1 cells with CD73+CD4+ T cells further contributes to the suppressive activity of these cells [131]. The CD39/CD73 axis is also essential for efficient activation, adhesion, and chemotaxis of neutrophils, which have been reported to have tumor promoting functions [24]. The majority of human Tregs are CD39+ but CD73−, however; CD73 in these cells is abundantly present in the intracytoplasmic granules [131]. The expression and activity of CD73 on Tregs is enhanced following TCR engagement [131].
7.3.2.3 Adenosine Binding P1 Receptors: A1, A2a, A2b, and A3
P1Rs are widely expressed by immune cells of both the myeloid and lymphoid lineage in mouse and humans (Table 7.3). Amongst the P1 receptors, A1R and A2BR expression is more restricted to myeloid cells, while A2AR and A3R are more ubiquitously expressed on both myeloid and lymphocyte populations [19, 96].
Expression of A1R and A3R on immune cells primarily activates anti-tumor immune responses. For example, pre-treatment of mice with CPA, an A1R agonist resulted in enhanced proliferation of splenic CD4+ T cells and increased production of IL-2 [153]. Similarly, activation of A3R using the agonist Cl-IB-MECA, enhanced NK cell functions, and improved anti-tumor immunity in mice [154].
The anti-inflammatory role of A2AR was first demonstrated by Sitkovky’s group in 2001. In their study, A2AR-deficient mice demonstrated exacerbated hepatitis and more severe tissue damage compared to their wild type (WT) counterparts [155]. Currently, we understand that A2AR signaling in both myeloid and lymphoid cells can participate in mediating local immunosuppression in the TME (Fig. 7.2). A2AR signaling suppresses macrophage differentiation, maturation, and activation [156]. Furthermore, Cekic et al. showed that the expression of A2AR on myeloid cells can increase anti-inflammatory IL-10 cytokine production by TAM and tumor-associated DC (TADC) in the TME. Additionally, conditional deletion of A2AR on myeloid cells significantly enhanced infiltration, activation, and effector functions of CD8+ T cells and NK cells in the tumor [12]. Likewise, the activation of A2AR on NK cells inhibits their ability to produce IFN-γ [157], while activation with A2AR agonist CGS21680 increased the proportions of Tregs in mixed lymphocyte cultures [158].
A2BR stimulation also promotes immunosuppression, but primarily on myeloid cells. Specifically, A2BR inhibits the differentiation of hematopoietic progenitor cells into mature myeloid cells and results in the increased accumulation of immature cells. These cells exhibit immunosuppressive functions through the production of vascular endothelial growth factor (VEGF), IL-8, TGF-β, and indolamine 2, 3-dioxygenase (IDO) [13]. Similarly, treatment of mice with an A2BR agonist, Bay60-6583 increased the frequencies of myeloid-derived suppressor cells (MDSC) infiltrating the B16F10 melanoma primary tumor, while depletion of these suppressor cells promoted anti-tumor effects in these mice, thus implying that A2BR signaling on MDSC promotes tumor progression [159].
7.4 Pre-clinical Evidence for Purinergic Signaling in Cancer
7.4.1 Extracellular ATP in Cancer
Given the extremely high concentration of extracellular ATP within the TME coupled with increased expression of P2 receptors on tumors and immune cells, it is not surprising that the purinergic pathway plays important roles in directly modulating tumor growth and anti-tumor immune responses. Interestingly, the P2X7R can have direct and opposite effects on cancer growth. Several lines of studies indicate that the engagement of P2X7R directly on tumor cells potentiates their growth whereas the same engagement on host cells such as DC, macrophages, and T cells activate potent anti-tumor responses. The pro-tumor effects of P2X7R include stimulating tumor cell growth, release of immunosuppressive factors and cytokines by MDSC, as well as stimulation of factors including VEGF to mediate tumor neovascularization [146]. To this end, P2X7R silencing in prostate cancer cells down-modulates genes that are involved in epithelial/mesenchymal transition including Snail, E-cadherin, Claudin-1, IL-8, and matrix-metalloproteases (MMP-3) [33]. Similarly, mice inoculated with mouse melanoma or human neuroblastoma cells and knocked down for P2X7R had slower tumor growth and significant reduction in metastases [33]. Inhibition of P2X7R using the selective inhibitor, AZ10606120 also caused a strong inhibition of tumor growth and reduction in VEGF production in B16F10 inoculated mice, compared to untreated controls [160].
By contrast to the above-mentioned studies, activation of P2X7R on DCs can promote anti-tumor activity. P2X7R has long been known for its role in the activation of the NLRP3 inflammasome and IL-1β release. Virgilio’s group demonstrated that P2X7R knockout mice exhibited accelerated B16F10 melanoma growth and lung metastases over their WT littermates. These effects were attributed, in part, to the low intratumor IL-1β that led to inefficient recruitment and infiltration of anti-tumor immune cells into the tumor [161]. Thereafter, studies confirmed that in the TME, the activation of NLRP3 inflammasome in DC through P2X7R positively influenced its antigen presenting capacity, and enhanced anti-tumor immunity in mice [162]. Additionally, IL-1β production through the inflammasome axis can, in conjunction with IL-23, promote IL-17 release by γδ T cells. This further promotes the release of IFN-γ by CD8+ T cells [162, 163]. Importantly, binding of ATP to P2X7R or P2Y2R triggers an immunogenic signal such that dying cancer cells are able to exert a potent anticancer vaccine effect [164]. This was elegantly demonstrated by Zitvogel and Kroemer, where they showed that chemotherapeutic drugs such as mitoxantrone or oxaliplatin induce release of ATP from dying tumor cells. This resulted in an enhanced infiltration of DC and T cells, which promoted potent anti-tumor immunity. Importantly, this immunogenic cell death (ICD) was abrogated in mice deficient in P2X7R and these mice were unable to mount tumor-specific CD8+ T cell responses [162]. Similarly, clinical observations corroborated this finding whereby loss-of-function polymorphism in P2X7R negatively affected disease outcome in breast cancer patients undergoing treatment with anthracycline-based chemotherapy [162].
Similar to P2X7R, P2Y2R can have direct pro-tumor effects. P2Y2R activation on MDA-MB-231 breast cancer cells increased their proliferation, ability to adhere to endothelial cells, and their production of MMP-9 and VEGF [92]. This study further showed that knockdown of P2Y2R suppressed invasion and migration of prostate cancer cells [165]. Interestingly, patients with NSCLC harboring mutations in the anaplastic lymphoma kinase (ALK) are often treated with ALK inhibitors, such as crizotinib. The overall response rate in these patients was 74% and progression free survival was a median of 10.9 months. Despite initial responses, resistance to crizotinib occurs in the majority of these patients [166]. Crizotinib resistant-NSCLC cell line, H3122 demonstrated an increased expression of the P2YR subtypes (P2Y1R, P2Y2R, and P2Y6R). These P2YRs upon activation led to increased protein kinase C (PKC) activity and production of endothelial growth factor receptor (EGFR) [167]. Overall, these findings suggest that P2YR inhibitors could be putative anticancer drug in the treatment of ALK-dependent NSCLC patients who develop resistance to ALK inhibitors.
7.4.1.1 Adenosine Generating Ecto-nucleotidases in Cancer Progression
Feng et al. demonstrated a role for CD39 in promoting tumor growth using mice lacking CD39. In this study, B16F10 melanoma growth and metastasis were markedly reduced in CD39 knockout mice compared with their corresponding WT littermates. In addition, endothelial cells in the CD39 knockout mice were impaired and were unable to influence tumor cell proliferation [72]. A similar observation was also reported by Sun et al. where they showed loss of host CD39 abrogated experimental liver metastases of B16F10 melanomas and MC38 colon tumors. Mechanistically, they showed using elegant adoptive T cell transfer setup experiments that CD39 expression on Tregs was a critical determinant in tumor rejection. Particularly, transfer of Tregs from WT donors into host CD39 KO mice but not vice versa resulted in the suppression of NK cell cytotoxic functions. This study also provided proof-of-principle that targeting CD39 activity could ameliorate tumor growth and metastases. Indeed, POM-1, a pharmacological inhibitor for CD39, prevented B16 melanoma and MC38 colon tumor growth. Importantly, the anti-tumor effect of POM-1 was similar to those observed with the CD39 knockout mice [168]. Given the short half-life of POM-1 in the circulation, several groups have now developed CD39-blocking monoclonal antibodies (mAbs). Human anti-CD39 mAb (clone 9-8B) has shown promising activity in inhibiting the enzymatic function of CD39 and improving survival in a lethal metastatic patient-derived sarcoma xenograft model [169]. Similarly, Bastid and co-workers recently described another anti-human CD39 mAb (clone BY40) that showed inhibition of CD39 enzymatic activity in SK-MEL5 human melanoma cells. Addition of this mAb to SK-MEL5 and T cell co-cultures resulted in efficient blocking of CD39 activity and increased the proliferation of CD4+ and CD8+ T cells. The therapeutic efficacy of this anti-CD39 mAb, however, remains to be tested in vivo [42]. In mice overexpressing CD39 and challenged with MC-26 colon cancer, significantly larger metastatic tumors compared with WT or heterozygous controls were observed [170]. Thus, CD39-blocking antibodies are potentially useful in alleviating immunosuppression and restoring immunity to cancers. By contrast to these encouraging observations, aged CD39 deficient mice were reported to spontaneously develop hepatocellular carcinoma owing to high accumulation of ATP and mechanistic target of rapamycin (mTOR) signal activation [171].
Similar to CD39, the importance of CD73 in promoting tumor growth through several host and tumor mediated pathways is also well understood. Using CD73 knockout mice, Stagg et al. demonstrated that the loss of host CD73 conferred resistance to B16F10 primary subcutaneous growth and experimental lung metastases. Such protection in the absence of CD73 was also seen with other tumor models including MC38 colon cancer, EG7 lymphoma, AT-3 mammary, EL4 lymphoma, and ID8 ovarian tumors. The effect of host CD73 loss was found to be dependent on the infiltration and activation of CD8+ T cells [14]. Additionally, two independent studies in 2011 observed that Tregs from CD73 knockout mice compared to WT mice were functionally inefficient in suppressing IFN-γ production from effector T cells [14, 172]. In the transgenic adenocarcinoma mouse prostate (TRAMP) model, absence of host CD73 protected these mice from de novo prostate cancer development; IFN-γ, NK cells, and CD8+ T cells were required for this protective effect [173]. Interestingly, increased protection to primary tumor was seen with the EG7 lymphoma model (over the parental EL4 lymphoma) and B16F10-SIY (over the parental B16F10) when inoculated into CD73 deficient mice, indicating a role for tumor immunogenicity in mediating optimal anti-tumor effects [172]. By contrast, the anti-metastatic effect seen in CD73-deficient mice is mediated independently of hematopoietic cells and possibly through endothelial cells. In vivo pharmacological blockade of CD73 with a selective inhibitor, APCP, or anti-CD73 mAb (clone TY/23) significantly reduced lung metastases [14]. Terp et al. demonstrated that anti-human CD73 mAb (cloneAD2) reduced the number of human breast cancer MDA-MB-231 and LM3 metastases by inducing the internalization of surface CD73. Importantly, the anti-metastatic effect was independent of CD73 catalytic function [46]. Furthermore, we now understand that the optimal protection to metastasis by the anti-CD73 mAb is largely dependent on the activation of Fc receptors by the host and the expression of CD73 on tumors, as recently demonstrated by Young and colleagues [45].
Related posts:
 Peptide-Based Therapeutic Cancer Vaccines
Peptide-Based Therapeutic Cancer Vaccines
 Current Status of Immuno-Oncology in Hematologic Cancers
Current Status of Immuno-Oncology in Hematologic Cancers
 Synergy Between Radiotherapy and Immunotherapy
Synergy Between Radiotherapy and Immunotherapy
 Toward Engineered Cells as Transformational and Broadly Available Medicines for the Treatment of Cancer
Toward Engineered Cells as Transformational and Broadly Available Medicines for the Treatment of Cancer
 Plasmacytoid DC/Regulatory T Cell Interactions at the Center of an Immunosuppressive Network in Breast and Ovarian Tumors
Plasmacytoid DC/Regulatory T Cell Interactions at the Center of an Immunosuppressive Network in Breast and Ovarian Tumors
 Immune Monitoring of Blood and Tumor Microenvironment
Immune Monitoring of Blood and Tumor Microenvironment
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


