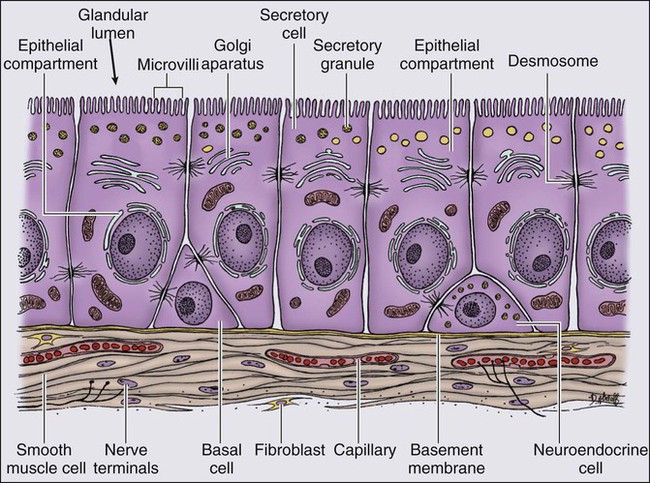
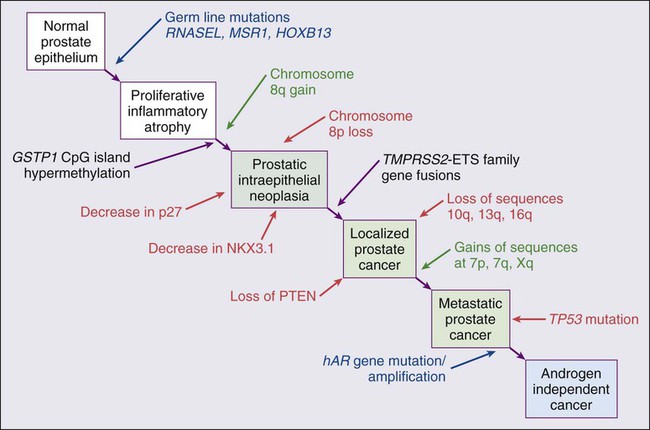
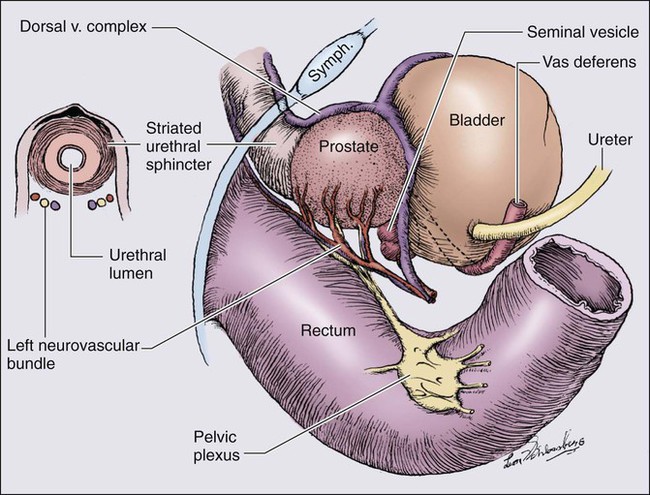
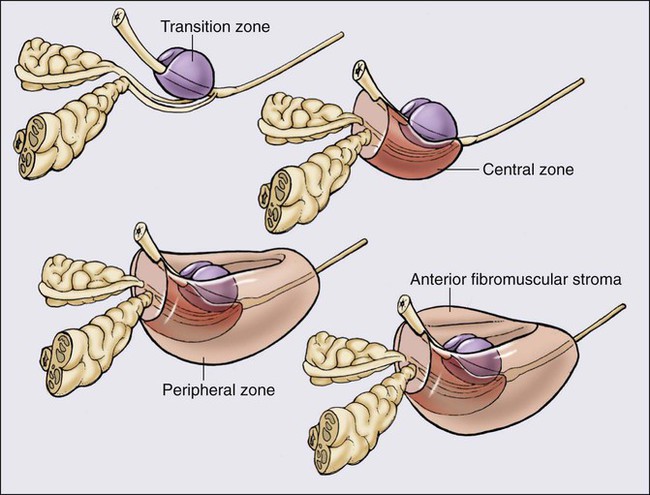
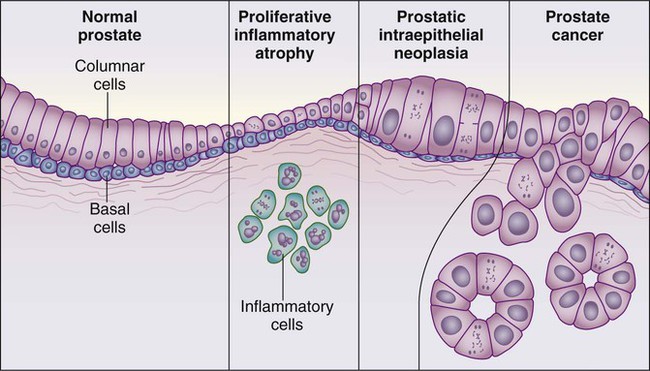
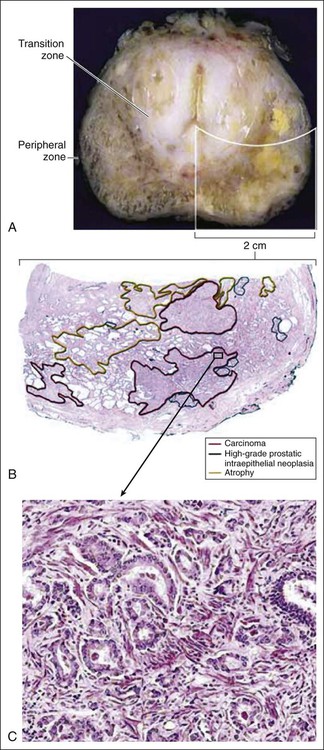
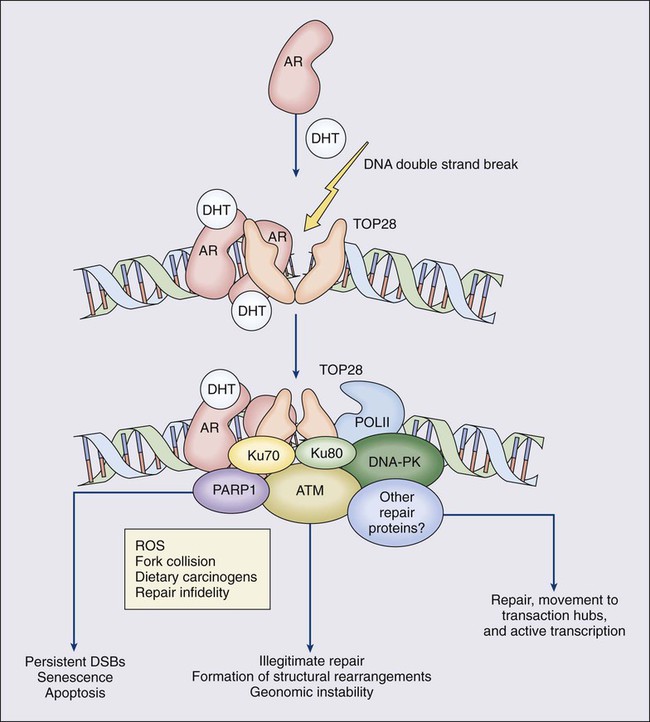
84 William G. Nelson, H. Ballentine Carter, Theodore L. DeWeese, Emmanuel S. Antonarakis and Mario A. Eisenberger • Prostate cancer is the most commonly diagnosed life-threatening cancer in men (241,740 cases and 28,170 deaths in 2012). • Small prostate cancers are present in 29% of men between ages 30 and 40 and 64% of men between ages 60 and 70. • The lifetime risk of a prostate cancer diagnosis is 1 in 6, and the risk of dying from prostate cancer is 1 in 35. • Age, family history, diet and lifestyle, and ethnicity are risk factors for prostate cancer development. • Germline mutations in RNASEL and MSR1, encoding proteins that function in host responses to infection, appear responsible for some cases of hereditary prostate cancer. • An inflammatory lesion, termed proliferative inflammatory atrophy (PIA), is an early precursor to prostate cancer. • Somatic inactivation of GSTP1, encoding a carcinogen-detoxification enzyme, may initiate prostatic carcinogenesis by increasing the vulnerability of prostate cells to damage mediated by oxidant and electrophilic carcinogens. • Gene fusions, involving TMPRSS2 and ETS family transcription factor genes, may contribute to the androgen dependence of prostate cancers. • Defects in the functions of NKX3.1, PTEN, and CDKN1B are common in prostate cancer cells. Screening, Diagnosis, and Staging • Prostate cancer screening using specific antigen (PSA) testing reduces the risk of prostate cancer death but may also lead to overdiagnosis of non-life-threatening disease. • Transrectal ultrasound (TRUS)-guided core needle biopsies are used to diagnose prostate cancer. • Stage, histologic grade (Gleason score), and serum PSA levels are prognostic factors. • Management options include observational strategies (watchful waiting and active surveillance), anatomic radical prostatectomy (with or without robot-assisted laparoscopic techniques), external beam radiation therapy, and brachytherapy. • A progressive rise in the serum PSA after treatment indicates prostate cancer recurrence. • Depending on the approach used, side effects associated with treatment of localized prostate cancer can include urinary, bowel, and sexual dysfunction. • Salvage therapy for prostate cancer recurrences after initial treatment include external radiation after surgery, or include surgery, brachytherapy, or cryosurgery after external beam radiation. • Adjuvant androgen suppression can improve survival for some men with prostate cancer treated with external beam radiation therapy. • Adjuvant external radiation improves survival for some men treated with radical prostatectomy. • Androgen suppression, most often accomplished via the use of luteinizing hormone–releasing hormone (LHRH) analogs or antagonists, with or without antiandrogens, is the most commonly used treatment. • Side effects can include loss of libido, hot flashes, gynecomastia, loss of lean muscle mass and bone density, and the development of metabolic syndrome. • Docetaxel and cabazitaxel chemotherapy improves the survival of men with progressive androgen-independent prostate cancer. • Second-line treatments targeting the androgen-signaling pathway, including abiraterone acetate and enzalutamide, prolong survival of men previously treated with androgen suppression and taxane chemotherapy. • Bisphosphonates and denosumab antagonize loss of bone density accompanying androgen deprivation, and reduce skeletal complications associated with metastatic prostate cancer progression. • Sipuleucel-T, a dendritic cell vaccine, has shown a survival benefit in men with advanced prostate cancer. Other immunotherapies are under development in clinical trials. In 2012, an estimated 241,740 prostate cancer diagnoses will be made in the United States, accompanied by an estimated 28,170 prostate cancer deaths.1 Beginning around 1994-1996, with widespread use of serum prostate-specific antigen (PSA) testing and digital rectal examination (DRE) for prostate cancer screening, and with increased treatment of clinically localized prostate cancer with surgery or radiation therapy, age-adjusted prostate cancer death rates have fallen steadily. Although this trend might indicate a beneficial impact of prostate cancer screening and/or early prostate cancer treatment on prostate cancer mortality, mass screening of the general population for prostate cancer remains controversial.2 One challenge for prostate cancer screening is the prevalence of the disease in the United States: autopsy series have revealed small prostate cancers in as many as 29% of men between ages 30 and 40 and 64% of men between ages 60 and 70.3 Obviously, not all of these men are at risk for symptomatic or life-threatening prostate cancer progression. In fact, many such men, if diagnosed with prostate cancer, may be at greater risk for treatment-associated morbidity. Currently, for U.S. men, the lifetime risk of a diagnosis of prostate cancer is about 1 in 6, whereas the lifetime risk of death from prostate cancer is on the order of 1 in 35.1 Over the past two decades, treatment approaches for men with prostate cancer have changed dramatically, with improvement in established prostate cancer treatments and the introduction of new prostate cancer treatment approaches. Now, men diagnosed with prostate cancer often face a bewildering array of treatment choices. Clearly, the physicians that care for these men must weigh the risks of prostate cancer progression against the potential for side effects from treatment, in the context of other health risks and life choices, to best use the current collection of treatments for the greatest benefit. The prostate is a male sex accessory gland that surrounds the urethra and contributes secretions to the ejaculate (Figure 84-1). Located in the pelvis, the prostate sits adjacent to the bladder and rectum, is surrounded incompletely by a thin capsule composed of collagen, elastin, and smooth muscle, and at the apex of the gland, forms part of the urethral sphincter apparatus.4 Nerves to the corpora cavernosa of the penis, needed for penile erection, travel through fascia along the posterolateral surface of the prostate. These nerves can be recognized as a neurovascular bundle by urologists and preserved during radical prostatectomy to minimize sexual dysfunction postoperatively.5,6 The prostate parenchyma has been divided into three zones that can be seen by transrectal ultrasonography, and recognized readily by surgical pathologists examining radical prostatectomy specimens: a central zone, surrounding the ejaculatory ducts and accounting for some 25% of the prostate; a transition zone, near the prostatic urethra with 10% of prostate tissue normally; and a peripheral zone, with the bulk of prostate tissue encompassing the posterolateral region of the prostate (Figure 84-2).7,8 The prostate requires androgenic hormones, and an intact androgen receptor, for normal growth and development. In the prostate, the major circulating androgenic hormone, testosterone, produced by Leydig cells in the testes upon stimulation by luteinizing hormone (LH), is converted by 5α-reductase (nicotinamide-adenine dinucleotide phosphate-dependent δ4-3-ketosteroid 5α-oxidoreductase) to 5α-dihydrotestosterone (DHT).9 DHT, a more potent androgen than testosterone, binds to intracellular androgen receptors, alters androgen receptor conformation to promote dissociation from chaperone proteins, triggers androgen receptor dimerization and transport into the cell nucleus, and activates the expression of selected target genes.10 Stereotypically, androgen receptor target genes are characterized by the presence of androgen response element (ARE) DNA sequences within the transcriptional regulatory region, permitting direct binding and trans-activation by the androgen receptor.11 For genes like KLK3 (encoding PSA), which are activated by the androgen receptor selectively in prostate cells, and not in cells of other tissues, the transcriptional regulatory region also contains additional DNA sequences (prostate-specific enhancer or PSE) conferring prostate-specific expression.12 The normal prostate epithelium is composed of basal epithelial cells, characterized by the expression of cytokeratins K5 and K14, and p63, columnar secretory epithelial cells, which express the androgen receptor, PSA, cytokeratins K8 and K18, prostate-specific membrane antigen (PSMA), and prostate-specific acid phosphatase (PAP); and rare neuroendocrine cells, that secrete chromogranin A, neuron-specific enolase, and synaptophysin (Figure 84-3).13 The basal epithelial cell compartment likely contains pluripotent prostatic stem cells, capable of self-renewal proliferation and of differentiation. In contrast, columnar secretory cells, specialized to produce secretions for the ejaculate, are terminally differentiated, particularly under the influence of androgenic hormones. The prostate epithelium is in turn supported by a stroma containing fibroblasts, smooth muscle cells, nerves, and blood vessels. Stromal cells, which also express the androgen receptor, secrete polypeptide growth factors, such as keratinocyte growth factor (KGF), that contribute to the regulation of epithelial homeostasis via a paracrine signaling mechanism.14,15 Abnormal stromal–epithelial interactions, with disordered regulation of epithelial cell proliferation and differentiation, may contribute to the pathogenesis of both prostate cancer and BPH.16 Prostate cancer cells, and PIN cells, arise from the prostatic epithelium. Even though transformed, such cells typically retain many of the phenotypic attributes characteristic of differentiated columnar secretory cells, including the expression of androgen receptor, PSA, PSMA, and PAP. Prostate cancers reminiscent of basal epithelial cells are exceptionally rare; prostate cancers with features of neuroendocrine cells are somewhat more common.17 However, unlike normal columnar epithelial cells, neoplastic prostate epithelial cells are capable of proliferation. This has led to the concept that the target cell for neoplastic transformation in the prostate may be an “intermediate” cell, in transit from a basal epithelial stem cell to a differentiated columnar secretory epithelial cell, with properties of both stem cells and differentiated cells.18,19 Another feature of neoplastic prostate epithelial cells, as compared with normal basal or columnar secretory cells, is that the neoplastic cells appear to use androgen receptor signaling not only for differentiation, but also for proliferation, as most prostate cancer cells tend, at least initially, to display some dependence on androgens for maintenance of growth and survival. Somatic fusions between an androgen-regulated gene, TMPRSS2, at chromosome 21q22, and genes encoding members of the ETS family of transcription factors, commonly found in prostate cancers, may provide a mechanistic explanation by which androgen signaling can promote prostate cancer cell growth.20,21 Ultimately, in life-threatening prostate cancer, prostate cancer cells escape from the prostate gland, proliferate in lymph nodes, in bones, and in other organs and become less and less dependent on androgenic hormones. Familial clusters of prostate cancer have been recognized since at least 1956, when Morganti et al. reported that men with prostate cancer were more likely to have relatives with prostate cancer than men without a prostate cancer diagnosis.22 In a study conducted more than three decades later, when detailed family histories were collected from men with prostate cancer and their spouses, the men with prostate cancer were more likely to have a brother or father with prostate cancer.23 Twin studies, comparing the tendency for concordant prostate cancer development between monozygotic twins, sharing all of their genes, and dizygotic twins, sharing half of their genes, have also hinted at a significant contribution of hereditary to prostate cancer: in a study of 44,788 pairs of twins in Sweden, Denmark, and Finland,24 42% of the prostate cancer cases (with a 95% confidence interval of 29% to 50%) were attributed to heredity. In principle, familial clustering of prostate cancer cases could be a result of inherited susceptibility genes, shared exposure to carcinogenic stresses, or to some sort of detection or diagnosis bias (e.g., the brother of a man diagnosed with prostate cancer may be more likely to pursue screening for prostate cancer). To discover the genetic contributions to prostate cancer in spite of these potential sources of bias, both linkage analyses of genetic loci in high-risk prostate cancer families and genomewide association studies (GWAS) of prostate cancer susceptibility in large populations have been conducted. Each approach has generated a number of candidate genes or gene regions (as many as 30 or more), with several in common, confirming the contribution of heredity to prostate cancer risk but underscoring the complexity of inherited prostate cancer susceptibility.25 Among the growing number of prostate cancer susceptibility genes discovered by mapping studies are RNASEL and MSR1.26,27 RNASEL encodes a latent endoribonuclease component of an interferon-inducible 2′,5′-oligoadenylate-dependent RNA decay pathway that functions to degrade viral and cellular RNA upon viral infection.28 MSR1 encodes subunits of a trimeric class A macrophage scavenger receptor capable of binding bacterial lipopolysaccharide and lipoteichoic acid, and oxidized high and low density serum lipoproteins (oxidized HDL and LDL).29 For both RNASEL and MSR1, not only have mutations been linked to prostate cancer susceptibility in families but variant alleles have been predicted to account for as many as 13% and 3% of sporadic prostate cancer cases, respectively.27,30,31 The identification of RNASEL and MSR1 as candidate prostate cancer susceptibility genes has intensified interest in the possibility that infection and/or inflammation might contribute to the pathogenesis of human prostate cancer. In mice, targeted disruption of RnaseL leads to diminished interferon-α activity and increased susceptibility to viral infection,32 whereas targeted disruption of Msr-A leads to increased vulnerability to infection with Listeria monocytogenes, Staphylococcus aureus, Escherichia coli, and Herpes simplex virus type 1.29,33–35 Further genetic support for this etiologic mechanism has come from analyses of common variants of other genes encoding participants in host inflammatory responses, including TLR4 and other members of toll-like receptor signaling pathways, MIC-1, IL1-RN, and COX-2, have also been associated with prostate cancer risk.36–40 An increased risk for prostate cancer has long been known for men in breast cancer families carrying BRCA2 mutations, characterized by disease with an aggressive natural history arising before age 55 years.41 Nonetheless, a role for BRCA2 genotyping in general prostate cancer practice has not been established. However, as evidence has accumulated that several additional germline DNA sequence variants may be associated with prostate cancer, the possibility that genetic testing might be used to aid in prostate cancer screening, detection, diagnosis, or risk stratification has emerged. In one analysis, five such sequence variants, three single nucleotide polymorphisms (SNPs) at 8q24 and one each at 17q12 and 17q24.3, were found to have a marked association with prostate cancer, especially for men with a family history of the disease, showing a 9.46-fold increased risk (with 95% confidence interval of 3.62 to 24.72) for a prostate cancer diagnosis.42 Accumulated epidemiological evidence implicates the environment as the major contributor to the development of most prostate cancers. Prostate cancer incidence and mortality display wide geographic variation, with high rates of prostate cancer incidence and mortality in the United States and Western Europe, and low prostate cancer risk more characteristic of Asia.43 African Americans in the United States have very high prostate cancer risk.44 The geographic variation in prostate cancer incidence and mortality can best be explained by lifestyle influences, because Asian immigrants to North America typically adopt a higher prostate cancer risk.47–47 The key aspect of lifestyle in the United States most likely responsible for high prostate cancer incidence and mortality is the diet, generally rich in animal fats and meats and poor in fruits and vegetables. In the Health Professions Follow-up Study, a prospective cohort study involving 51,529 men, total fat intake, animal fat intake, and consumption of red meats were associated with increased risks of prostate cancer development.48 Red meat consumption was similarly correlated with prostate cancer risks in the Physicians Health Study49 and in a large cohort study in Hawaii.50 The cooking of red meats at high temperatures, or on charcoal grills, is known to lead to the formation of both heterocyclic aromatic amine and polycyclic aromatic hydrocarbon carcinogens.51,52 Ingestion of 2-amino-1-methyl-6-phenylimidazopyridine (PhIP), one of the heterocyclic amine carcinogens that appear in “well-done” red meats, leads to prostate cancer in rats.53 Consumption of dairy products also appears to increase prostate cancer risk, an effect that may be more attributable to calcium intake than to dietary fat or protein.54 In contrast, adequate consumption of vegetables and antioxidant micronutrients is accompanied by reduced prostate cancer risk. Consumption of tomatoes, which contain lycopene, and of cruciferous vegetables, which contain sulforaphane, may protect against prostate cancer development.55,56 Antioxidant micronutrients, such as vitamin E and selenium, may reduce prostate cancer risk only when correcting dietary deficiencies: a large trial (SELECT) of supplementation with vitamin E and selenium to prevent prostate cancer failed to show a benefit.57 Chronic or recurrent inflammation is known to play a causative role in the development of many human cancers, including cancers of the liver, esophagus, stomach, large intestine, and bladder. Inflammatory changes have been recognized in prostate tissues for many years, leading to speculation that inflammation might contribute in some way to prostate cancer development.58 However, over the past few years, evidence has accumulated in support of a more critical role for prostatic inflammation in the pathogenesis of prostate cancer. Inflammatory changes are present in almost all radical prostatectomy specimens from men with prostate cancer. Because inflammation in the prostate is not usually associated with symptoms, the prevalence of prostate inflammation is not known, and the association with prostate cancer has been difficult to test.59,60 A syndrome of irritative voiding symptoms and pelvic pain, perhaps attributable to inflammation near the prostatic urethra, is reported by some 9% or more of men between 40 and 79 years of age, with as many as 50% of such men suffering more than one episode by age 80 years.61 Most episodes of symptomatic prostatitis are not clearly attributable to specific infectious agents. Even so, sexually transmitted infections do appear to increase prostate cancer risk.62,63 Nonetheless, if prostate infection and inflammation lead to prostate cancer, the mechanism does not appear likely to involve direct transformation of prostate epithelial cells by microbial DNA. Instead, the production of microbicidal oxidants by inflammatory cells, such as superoxide, nitric oxide, and peroxynitrite, may promote prostate cancer development by triggering cell and genome damage.64,65 Increased production of oxidants by inflammatory cells in the prostate may be why decreased prostate cancer risk has been associated with intake of a variety of antioxidants or of nonsteroidal antiinflammatory drugs, and why RNASEL and MSR1, two of the prostate cancer susceptibility genes identified thus far, encode proteins that function in host responses to infections. Despite these provocative hints, the contribution of prostate inflammation to prostatic carcinogenesis has been difficult to assess. However, in 1999, De Marzo et al. provided the most compelling linkage of prostate inflammation to prostate cancer by proposing that a prostate lesion, termed proliferative inflammatory atrophy (PIA), might be a precursor to PIN and to prostate cancer (Figure 84-4).66 Areas of the prostate, containing epithelial cells that do not fully differentiate into columnar secretory cells, have long been recognized as focal atrophy lesions by prostate pathologists.58,67 The term PIA has been used to describe those focal atrophy lesions that contain proliferating epithelial cells, are associated with chronic inflammation, and are often located adjacent to PIN lesions and/or prostate cancers.68 The epithelial cells in PIA lesions typically express high levels of stress-response polypeptides such as GSTP1, GSTA1, and cyclooxygenase 2 (COX-2). Loss of GSTP1 expression in rare PIA lesions, attributable to de novo GSTP1 CpG island hypermethylation, may be what leads to the development of PIN and prostate cancer.69 Prostate cancer cells typically contain a plethora of somatic genome alterations, including gene mutations, gene deletions, gene amplifications, chromosomal rearrangements, and changes in DNA methylation (Figure 84-5). In the United States, prostate cancer diagnoses are typically made in men between 60 and 70 years of age, whereas small prostate cancers have been detected at autopsy in nearly 30% of men between 30 and 40 years of age.3 Thus, the somatic genome changes present in prostate cancers have often accumulated over many decades. The acquisition of somatic genome changes in the prostate may be influenced by lifestyle as well: although small prostate cancers have been detected at autopsy in men from geographic regions with low prostate cancer mortality, these small prostate cancers are usually only present in much older men.72–72 Also, in the United States, prostates removed at radical prostatectomy for prostate cancer usually contain more than one prostate cancer lesion (Figure 84-6). Over the years, several techniques have been used to catalog genome accidents in prostate cancer cells, including karyotyping, fluorescence in situ hybridization (FISH), comparative genome hybridization, loss of heterozygosity analyses, and genomewide microarray and/or sequencing approaches. In one such study, each prostate cancer case exhibited a mean of 3866 base mutations (range 3192 to 5865), 20 nonsilent coding sequence mutations (range 13 to 43), and 108 rearrangements (range 43 to 213).73 In another, DNA hypermethylation was found at 5408 regions of the genome, with 73% of the regions near genes (5′, 3′, or intron–exon junctions), and 27% of the regions at conserved intergenic sites.74 Often, these analyses reveal different chromosomal abnormalities in different cancer cases, in different cancer lesions in the same cancer case, and in different areas within the same cancer lesion. The propensity to develop such a heterogeneous collection of somatic genome lesions over so many years, and in a manner so sensitive to environment and lifestyle, suggests strongly that prostate cancers likely arise as a consequence of either chronic or recurrent exposure to genome-damaging stresses, defective protection against genome damage, or some combination of both processes. The resultant genomic instability may be the reason some prostate cancers progress to threaten life.75,76 One characteristic somatic genome alteration, a rearrangement, drives the production of fusion transcripts between an androgen-regulated gene, TMPRSS2, at chromosome 21q22, and members of the ETS family of transcription factors (Figure 84-7).20 Fusion partners for TMPRSS2 include ERG (also at chromosome 21q22), ETV1 (at chromosome 7p21), and ETV4 (at chromosome 17q21).20,77,78 The gene rearrangements may result from a mishap during the androgen receptor transcriptional trans-activation of TMPRSS2 in which tangling or untangling of DNA by TOP2B triggers DNA double-strand breaks that recombine with ETS family partner genes via nonhomologous end-joining (Figure 84-8).79,80 The appearance of the resultant fusion transcripts provides a plausible mechanism for the dependence of prostate cancer cells on androgenic hormones for growth and survival, as the expression of ETS family transcription factors can be stimulated by androgen action. TMPRSS2–ERG fusions have been detected in ~60% of prostate cancers and in >20% of PIN lesions.81 ERG is highly expressed by many prostate cancers, and not at all by others, though neither ERG expression nor the presence of ERG fusion transcripts appears to have great prognostic significance.81–84 Nonetheless, the presence of TMPRSS2-ERG fusion transcripts does seem to define a molecular subset of prostate cancer. Rarer somatic alterations, including SPOP mutations (6% to 15%) and SPINK1 overexpression (~10%), are restricted to cases devoid of TMPRSS2-ETS family rearrangement rearrangements.85,86 Hypermethylation of CpG island sequences encompassing the regulatory region of GSTP1, encoding the π–class glutathione S-transferase (GST) is the most common somatic genome change yet reported for prostate cancer.87,88 GSTs catalyze the detoxification of carcinogens, and of other reactive chemical species, via conjugation with the intracellular scavenger glutathione. In mice, targeted disruption of π-class GST genes leads to increased skin tumors after treatment with the carcinogen 7,12-dimethylbenzanthracene (DMBA).90 Similarly, human prostate cancer cells devoid of GSTP1 appear especially vulnerable to genome damage mediated by exposure to N-OH-PhIP, the charred meat carcinogen that causes prostate cancer when fed to rats, and by exposure to oxidant stresses.91 In the normal prostate epithelium, GSTP1 is present at high levels in basal cells, and in lower levels in columnar secretory cells, though the enzyme can be induced in columnar epithelial cells subjected to genome-damaging stresses. In contrast, the enzyme is almost never present in prostate cancer cells. In nearly all cases, the absence of GSTP1 expression in prostate cancer cells can be attributed to hypermethylation of GSTP1 CpG island sequences, a somatic genome change that prevents GSTP1 transcription. Absence of GSTP1 expression and GSTP1 CpG island hypermethylation may also be characteristic of cells comprising PIN lesions, thought to be precursors to prostate cancer.92 The mechanism by which hypermethylated GSTP1 CpG island alleles arise during prostatic carcinogenesis remains to be elucidated. Nonetheless, prostate cells carrying inactivated GSTP1 genes appear to enjoy some sort of selective growth advantage early during the development of prostate cancer. NKX3.1 encodes a prostate-specific homeobox gene essential for normal prostate development that may be a target for somatic loss on chromosome 8p21.93 NKX3.1 has been shown to bind DNA and to repress PSA expression via interactions with ETS transcription factors.94,95 Mice carrying one or two disrupted Nkx3.1 alleles manifest prostatic epithelial hyperplasia and dysplasia.96,97 In men, loss of 8p21 DNA sequences occurs early during prostatic carcinogenesis, with 63% of PIN lesions and >90% of prostate cancers, showing loss of heterozygosity at polymorphic 8p21 marker sequences in one report.98 However, although mapping studies have indicated that NKX3.1 lies within a common region of deletion, encompassing 2 megabases at 8p21, molecular pathology analyses have not yet established NKX3.1 as a somatic target for inactivation during prostatic carcinogenesis because somatic NKX3.1 mutations have not been identified. Nonetheless, loss of NKX3.1 expression does appear to accompany prostate cancer progression. PTEN, a tumor suppressor gene encoding a phosphatase active against both proteins and lipid substrates, appears to be a common target for somatic alteration during prostate cancer progression.99–106 PTEN is an inhibitor of the phosphatidylinositol 3′-kinase/protein kinase B (PI3K/Akt) signaling pathway needed for cell cycle progression and cell survival. Although PTEN is expressed by normal prostate epithelial cells, and by cells present in PIN lesions, the expression of PTEN is often diminished in prostate cancers, with many prostate cancers containing collections of neoplastic cells with no PTEN.107 PTEN defects have been found in a wide variety of cancers and cancer cell lines.101 For prostate cancer, a number of somatic PTEN alterations have been reported, including homozygous deletions, loss of heterozygosity, mutations, and probable CpG island hypermethylation. However, despite common losses of 10q sequences near PTEN in prostate cancers, somatic mutations at the remaining PTEN alleles are not as frequent. In a study of prostate cancer metastases recovered at autopsy, somatic PTEN alterations were even more common than in primary prostate cancers, and a significant heterogeneity in PTEN defects in different metastatic deposits from the same patient was also evident.106 Haploinsufficiency for PTEN may contribute to the phenotype of transformed cells in the prostate. Pten+/– mice display prostatic hyperplasia and dysplasia, and crosses of Pten+/– mice with Nkx3.1+/– mice have revealed that Pten+/–Nkx3.1+/– mice and Pten+/–Nkx3.1–/– mice develop lesions reminiscent of human PIN.110–110 Defective regulation of p27, a cyclin-dependent kinase inhibitor encoded by CDKN1B, may also accompany prostatic carcinogenesis.111,112 In PIN cells and prostate cancer cells, p27 levels are almost always diminished, though the mechanism(s) for the reduction in p27 levels appear complex: somatic loss of DNA CDKN1B sequences at 12p12-13 have been reported for only 23% of localized prostate cancers, 30% of prostate cancer lymph node metastases, and 47% of distant prostate cancer metastases.113 In place of CDKN1B gene alterations, p27 polypeptide levels may be lowered indirectly by inadequate PTEN repression of the PI3K/Akt signaling pathway.116–116 In this way, low p27 levels may be as much a result of loss of PTEN function as of CDKN1B alterations. The critical contribution of PTEN to epithelial growth regulation in the prostate is evident in mice, where disruption of Cdkn1b alleles leads to prostatic hyperplasia, and Pten+/–Cdkn1b–/– mice develop prostate cancer by 3 months of age.111,117 Metastatic prostate cancer is almost always treated with androgen deprivation, antiandrogens, or a combination of androgen deprivation and antiandrogens.118,119 However, despite such treatment, androgen-independent prostate cancer cells eventually emerge and progress to threaten life. Curiously, in these cells, androgen receptor expression and androgen receptor signaling remain intact despite the absence of androgens.120,121 Somatic alterations of AR have been reported for many prostate cancers, especially for androgen-independent prostate cancers. AR amplification, accompanied by high-level expression of androgen receptors, may promote the growth of androgen-independent prostate cancer cells by increasing the sensitivity of the cells to low androgen levels.122 AR mutations, encoding androgen receptors with altered ligand specificity have also been detected; for some of the mutant androgen receptors, even antiandrogens can act as agonist ligands.125–125 When 44 mutant androgen receptors from prostate cancers were evaluated for transcriptional regulatory capabilities, 16% of the receptors had lost transcriptional activation activity, 45% of the receptors had gained some transcriptional regulatory ability, 32% of the receptors maintained some partial transcriptional modulatory activity, and the remaining 7% behaved like wild-type receptors.126 In addition to somatic AR gene changes, androgen-independent prostate cancer cells with wild-type androgen receptors may activate androgen receptor signaling even in the absence of androgens, via posttranslational modifications of the androgen receptor and/or androgen receptor coactivators in response to other growth factor signaling pathways.120,127–130 Alterations in gene expression in prostate cancers have been catalogued using cDNA microarray technologies.131–142 Among the many genes exhibiting over- or underexpression in prostate cancers, the products of at least two genes appear consistently increased, and the product of a third gene appears to become elevated during androgen-independent progression. Hepsin, located at 19q11-13.2, encodes a transmembrane serine protease, expressed at high levels in many normal tissues.143 Hepsin may contribute to prostate cancer progression: forced overexpression of hepsin in mouse prostates leads to disorganization of the epithelial basement membrane and increased metastasis.144 α-Methylacyl-CoA racemase (AMACR), a mitochondrial and peroxisomal enzyme that acts on pristanoyl-CoA and C27-bile acyl-CoA substrates to catalyze the conversion of R– to S-stereoisomers in order to permit metabolism by β-oxidation, has been reported to be overexpressed in almost all prostate cancers.145,146 Germline AMACR mutations lead to adult-onset neuropathy.147 Immunohistochemistry studies, which have revealed that AMACR is occasionally present in normal prostate cells, increased in PIN cells, and further elevated in prostate cancer cells, have prompted the use of antibodies against AMACR as tools for prostate cancer diagnosis by surgical pathologists.146,148 The polycomb protein enhancer of zeste homolog 2 (EZH2), a transcriptional regulatory protein, is elevated in metastatic androgen-independent prostate cancer.149 The mechanism by which EZH2 contributes to prostate cancer progression has not been established. However, elevated EZH2 expression in primary prostate cancers portends a poor prognosis.149 Telomeres, containing repeat DNA sequences at the termini of chromosomes, protect against loss of chromosome sequences during genome replication. DNA ends tend to shorten each generation as a consequence of bidirectional DNA synthesis (the “end-replication” problem); the telomere repeat sequences serve as templates for the enzyme telomerase, which can extend the chromosome termini and maintain chromosome integrity through cell division.150 Growth dysregulation accompanying the development of most human cancers tends to lead to cell proliferation in the absence of telomerase, and to shortened chromosome telomeres.151 Critically shortened telomere sequences may promote genome instability by increasing illegitimate DNA recombination.152,153 Mice carrying disrupted genes needed for a functioning telomerase show increased numbers of cancers, especially when crossed to mice with defective p53 genes.154 In the prostate, short telomere repeat sequences appear characteristic of cells both in PIN lesions and in prostate cancer.157–157 At some point, most cancer cells activate the expression of telomerase, providing some maintenance of chromosome termini. Telomerase expression has been detected in prostate cancers, but not at high levels in normal prostate tissues or in BPH.155 In men with early-stage prostate cancers, physical findings, if present, are usually limited to an abnormal DRE, used for both screening and staging. Palpable areas of induration, or asymmetric firmness of the gland, suggest the presence of prostate cancer, but these findings can also be caused by prostate inflammation (especially granulomatous prostatitis), by benign prostatic hyperplasia (BPH), and by prostatic stones. DRE has only fair reproducibility in the hands of experienced examiners.158 When used alone for detection of prostate cancer, DRE misses from 23% to 45% of the cancers that are subsequently detected by prostate biopsies done for serum PSA elevations or for transrectal ultrasound (TRUS) abnormalities.161–161 In addition, prostate cancers detected by DRE are at an advanced pathological stage in more than 50% of men.162,163 The positive predictive value of DRE (the fraction of men who have prostate cancer if the DRE is abnormal) ranged from 4% to 11% in men with PSA levels from 0.0 to 2.9 ng/mL, and from 33% to 83% in men with PSA levels of 3.0 to 9.9 ng/mL or more.164 When DRE and PSA are used in prostate cancer screening, detection rates are higher with PSA than with DRE and highest with both tests together.165 Furthermore, a DRE abnormality tends to be associated with the presence of high-grade cancer.166 Thus DRE and PSA are generally considered to be complementary tests and a prostate biopsy is usually recommended for men with an abnormality on DRE that is suspicious for prostate cancer regardless of the PSA level. PSA is a member of the human kallikrein gene family of serine proteases encoded by KLK3 located on chromosome 19.167 A component of the ejaculate, PSA is produced by columnar secretory cells in the prostate. PSA expression is regulated by androgens, becoming detectable in serum at puberty accompanying increases in luteinizing hormone and testosterone. In the absence of prostate cancer, serum PSA levels increase with age and prostate volume and are generally higher in African Americans. Cross-sectional population data suggest that the serum PSA increases 4% per milliliter of prostate volume, and that 30% and 5% of the variance in PSA can be accounted for by prostate volume and age, respectively.168 Serum PSA elevations likely occur as a result of disruption of the normal prostate architecture, permitting PSA to diffuse into the prostate parenchyma and gain access to the circulation. This can occur in the setting of both benign and malignant prostate diseases (prostatitis, BPH, and prostate cancer) and as a result of prostate manipulation (prostate massage and prostate biopsy).169 Although the presence of some type of prostate disease is the most important determinant driving elevation of the serum PSA, an increased serum PSA is not specific for prostate cancer. Furthermore, not all men with prostate disease have elevated serum PSA levels. Treatments targeting the prostate gland (for BPH or for prostate cancer) can lower serum PSA by decreasing the number of prostatic epithelial cells capable of producing PSA, and by decreasing the amount of PSA produced by each cell. Modulation of sex steroid hormone levels for treatment of BPH or prostate cancer, radiation therapy for prostate cancer, and surgical ablation of prostate tissue for BPH or prostate cancer can all lead to decreases in serum PSA. 5α-Reductase inhibitors, like finasteride and dutasteride, lower PSA levels by 50% after 12 months of treatment.170 Thus for men treated with these agents for 12 months or more, the serum PSA level should be doubled to estimate the “true” PSA value. Interpretation of serum PSA values should always take into account the presence of prostate disease, previous diagnostic procedures, and prostate-targeted treatments. The serum PSA value, along with the findings at DRE, correlate directly with the risk of prostate cancer at biopsy (Table 84-1). Furthermore, the PSA level at a young age anticipates the future risk of being diagnosed with prostate cancer decades later (Figure 84-9). Gann et al. first showed that the risk of a prostate cancer diagnosis, including the diagnosis of life-threatening disease, incrementally and directly with PSA over the decade after a baseline measurement, even at low PSA levels (below 4.0 ng/mL); a finding that has now been confirmed by many others.173–173 These observations may allow a more targeted approach to prostate cancer screening using a baseline PSA value to direct screening intensity.174,175 Table 84-1 Positive Predictive Value of DRE and PSA in a Multicenter Screening Trial DRE, Digital rectal examination; PPV, positive predictive value; PSA, prostate-specific antigen. Data from Catalona WJ, Richie JP, Ahmann FR, et al. Comparison of digital rectal examination and serum prostate specific antigen in the early detection of prostate cancer: results of a multicenter clinical trial of 6,630 men. J Urol 1994;151:1283. In the early years of PSA testing, most clinicians used PSA as a dichotomous test (i.e., the PSA was “elevated” or not) with elevations triggering a prostate biopsy. However, data from the placebo arm of the Prostate Cancer Prevention Trial (PCPT176), which accrued men with a serum PSA value <3.0 ng/mL, has suggested that there is no PSA cut-off level with both high sensitivity and high specificity for prostate cancer, and that virtually no level is low enough to exclude the presence of prostate cancer. Instead, serum PSA tests may be more appropriately thought of as measures of a continuum of prostate cancer risk, with the risk of cancer (and of high-grade cancer) rising directly with the PSA level, an observation first made by Gann et al. using a cohort population study design.171
Prostate Cancer
Introduction
Prostate Anatomy and Function
Genetics and Epidemiology
Genetic Predisposition to Prostate Cancer
Epidemiology of Prostate Cancer
Prostate Inflammation and Prostate Cancer
Etiological and Biological Characteristics
Somatic Genome Alterations in Prostate Cancer Cells

Changes in Gene Expression in Prostate Cancers
Telomere Shortening During Prostatic Carcinogenesis
Prostate Cancer Screening, Early Detection, and Prevention
Clinical Evaluation
Digital Rectal Examination
Serum PSA
Serum PSA and Prostate Cancer Detection
DRE
PSA
PPV (%)
Abnormal
Any
21.4
Any
>4
31.5
4–10
26.1
>10
52.9
Normal
>4
24.4
Abnormal
<4
10.0
4–10
40.8
>10
69.1
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Prostate Cancer