Fig. 8.1
Milestones in senescence research: a timeline
8.1.1 Hallmarks of Cellular Senescence
Senescent cells are not characterized by a specific biomarker. Expression of a group of markers defines a senescent cell, however senescent cells not always express all the markers. Cell cycle growth arrest is a crucial mechanism for the identification of all types of senescence, both in vitro and in vivo. Nevertheless, as multiple cellular mechanisms can drive a stable replicative arrest in cells, cell cycle exit cannot be considered a unique marker for senescence. Senescent cells are commonly characterized by an increased cell size and a more flattened shape than normal cells and frequently exhibit an increased expression of senescence associated β-galactosidase activity (SA-β-gal) that in these cells can be detected at pH 6. Also augmented levels of p19ARF, p53, PAI-1 and of the cell cycle inhibitors p16INK4a, p21/CIP1 and p27 are generally ascribed as senescence biomarkers. In addition to this, senescence cells may exhibit senescence associated heterochromatin foci formation (SAHF), persistent DNA damage response (DDR) and commonly secrete large number of growth factors, cytokines and proteases, known as the senescence associated secretory phenotype (SASP). All the above-mentioned factors together define the hallmarks of senescence (Collado et al. 2007; Kuilman et al. 2010). Nevertheless, some of the cited senescence markers still need to be confirmed in vivo, and it is still necessary to clarify to what extend they can be considered valid across tissues and species.
8.1.2 Causes of Cellular Senescence
8.1.2.1 Telomere Shortening
Telomeres are regions of repetitive nucleotide sequences that protect the end of the chromosome from deterioration or from fusion with neighboring chromosomes. Telomeres shorten with each cell division (S phase) and each round of DNA replication leaves 50–200 bp of unreplicated DNA at the 3′ end. Telomerase adds bases to the ends of telomeres maintaining the function of these repeats. As cells divide repeatedly, there is not enough telomerase to compensate telomeres erosion, so the telomeres grow shorter and the cells age. Cancer cells escape senescence by activating telomerase enzyme and by other mechanisms, which prevent the telomeres from getting shorter (Shay 2014), thus allowing them to proliferate indefinitely. This makes telomerase an attractive target for the development of novel anti-cancer therapeutic agents (Wong et al. 2013). Telomeres are subject to attrition due to the fact that DNA polymerase fails to completely replicate the lagging strands. In the early 1970s, Olovnikov (1971) and Watson (1972) independently described this so-called “end replication problem”, which contributes to telomere shortening. Thus, telomeres act as a molecular clock, reflecting the replicative history of a primary cell (Harley 1990). When telomeres reach a critical minimal length, their protective structure is disrupted. This triggers a DNA damage response (DDR), which is associated with the appearance of foci that stain positive for γ-H2AX (a phosphorylated form of the histone variant H2AX) and the DDR proteins 53BP1, NBS1, and MDC1. Moreover, the DNA damage kinases ATM and ATR are activated in senescent cells (d’Adda di Fagagna et al. 2003). After amplification of the DDR signal, these kinases activate CHK1 and CHK2 kinases. Communication between DDR-associated factors and the cell cycle machinery is brought about by phosphorylation and activation of several cell cycle proteins, including CDC25 (a family of phosphatases) and p53. In addition to this, differential expression of p53 isoforms has been linked to replicative senescence (Fujita et al. 2009). Together, these changes can induce a transient proliferation arrest, allowing cells to repair their damages. However, telomere shortening does not completely explain the reason why mammalian cells age. Some mammals such as mice have longer telomeres that human telomeres and express higher telomerase levels (Kipling and Cooke 1990; Prowse and Greider 1995). However mice life span is significantly shorter that human life span. Elongation of cells life span can be achieved by culturing human cells in serum-free medium supplemented with a number of defined growth factors (Loo et al. 1987) or by culturing cells under physiological oxygen conditions (Parrinello et al. 2003). In contrast, oxidative stress induces senescence in cultured human cells (Packer and Fuehr 1977; Chen et al. 1995; Yuan et al. 1995). Thus, the immortalization of mammalian cells requires not only telomere maintenance, but also optimal culture conditions (Mathon et al. 2001; Ramirez et al. 2001; Tang et al. 2001; Herbert et al. 2002) (for review, see Wright and Shay (2002)). Oxidative stress-mediated DNA damage is also an important determinant of telomere shortening (Richter and von Zglinicki 2007). Zhou et al. (2014) have demonstrated that ER-stress transiently activates the catalytic components of telomerase (TERT), depletion of hTERT sensitizes cells to undergo apoptosis, whereas increased hTERT expression reduces ER stress-induced cell death independent of catalytically active enzyme or DNA damage signaling. Recently, it was also demonstrated that lifestyle modification like diet, activity, stress management, and social support have resulted in increased telomere length and telomerase activity (Ornish et al. 2013) (Fig. 8.2).
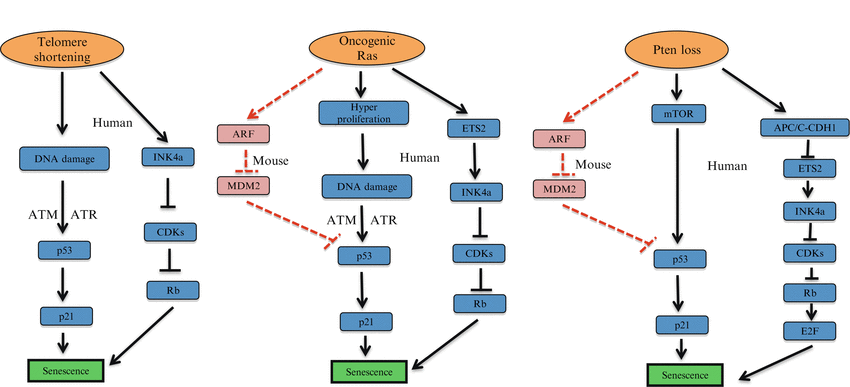
Fig. 8.2
Causes of cellular senescence. Telomere shortening, overexpression of specific oncogenes (e.g. Ras), and loss of tumor suppressor genes (e.g. Pten) as major mechanisms underlying cellular senescence
8.1.2.2 Stress-induced Cellular Senescence
Contrary to replicative senescence, stress induced senescence does not depend on telomere length. Extrinsic and intrinsic physiological stresses are reported to promote senescence in normal and cancer cells. Extrinsic stresses include treatment with DNA damage agents such as hydrogen peroxide (Chen and Ames 1994), UV- or γ- irradiation (Rodemann et al. 1989), tert-butylhydroperoxide (Toussaint et al. 1992) or different anti-cancer chemotherapy drugs (Sanchez-Prieto et al. 2000; Benhar et al. 2002). Intrinsic stresses include subjecting cells to abnormal O2 levels (von Zglinicki et al. 1995) and inadequate nutrients and culturing condition. This last phenomenon is also termed “culture shock” (Sherr and DePinho 2000), applies to both human and mouse cells and it is independent from telomere length. Oxidative stress, ER-stress and interferon-related responses, and signaling via either insulin growth factors (IGF) or mitogen activated protein kinases (MAPK), are various stress pathways that also cause cellular senescence (Campisi and d’Adda di Fagagna 2007). These stresses initiate various cellular signaling pathways, which turn to activate the p53 protein, the Rb protein, or both pathways. Distinguishing between replicative senescence and stress-induced senescence is very subtle, as these titles merely reflect the fact that a spectrum of different stimuli feed into one response program or other.
8.1.2.3 Overexpression of Oncogenes
Several oncogenes induce senescence both in vitro and in vivo. Oncogene induced senescence (OIS) is regulated by a complex signaling network that opposes the process of malignant tumor formation from benign tumors. OIS driven by HRASG12V or BRAF overexpression in human fibroblasts is triggered by massive hyperproliferation and DNA hyper-replication. This in turn sparks the activation of an S phase specific DDR that promote senescence (Di Micco et al. 2006). Although this DDR is initiated by a mechanism that is distinct from that of replicative senescence, it shares with the latter effectors and primary pathways and also results in the formation of senescence-associated DNA damage foci (SDF formation) (d’Adda di Fagagna 2008). Hyperproliferation and DDR are essential for OIS. Indeed OIS cannot be triggered in cells that lack DDR such as ATM/ATR deficient cells or in cells pre-treated with aphidicolin, a selective cell cycle inhibitor (Di Micco et al. 2006) (Fig. 8.2).
RAS
RAS was first identified in 1964 as a human oncogene and was described as a transforming retrovirus that produces tumors in mice. Mutations in oncogenic HRAS are most common in a wide variety of human cancers and are found in 30 % of the tumors (DeNicola et al. 2011). At least five inherited mutations in the HRAS gene have been identified in people with Cosetello syndrome. Each of these mutations changes a single amino acid in a critical region of the HRAS protein. The most common mutation replaces the amino acid glycine with the amino acid serine at position 12. Ras requires cooperation of another oncogene or inactivation of tumor suppressor genes like p53 and p16INK4a for transformation. Over expression of oncogenic HRAS in primary cells results in a permanent form of cell cycle arrest called oncogene induced senescence. Ras-induced senescence is accompanied by the upregulation of the tumor suppressors PML, p53 and p16INK4a (Serrano et al. 1997). Oncogene-induced senescence is executed via p53 and p16INK4a, which are involved in proliferation, differentiation and cell death. Inactivation of p53 and p16INK4a proteins results in bypassing Ras-induced senescence. This explains why Ras-induced senescence evolved as a protective barrier to prevent tumor progression (Courtois-Cox et al. 2008). p53 is an essential regulator of OIS (Ferbeyre et al. 2000; Wei et al. 2001), through transcriptional activation of target genes, in particular CDKN1A (encoding the protein p21/CIP1) (Serrano et al. 1997). In OIS, the activation of p53 is driven by phosphorylation (Di Micco et al. 2006), with concomitant stabilization of the protein mediated by ARF induction (Serrano et al. 1997). However, ARF induction may have a more limited role for the upregulation of p53 in a human context (Brookes et al. 2002). In addition to p53, OIS also engages other senescence effectors, including p16INK4a (for example, through ETS2 (Ohtani et al. 2001) and derepression of the genomic locus through inactivation of the polycomb group complex (Bracken et al. 2007). Moreover there are now several mouse models providing physiological evidence for OIS in vivo (Braig et al. 2005; Collado et al. 2005; Dankort et al. 2007; Sarkisian et al. 2007). Interestingly, the senescence response that is induced through the loss of the Rb1 tumour suppressor gene in vivo has also been reported to display OIS-like features that are mediated by activation of NRAS (Shamma et al. 2009). Transcriptional repression of pro-proliferative genes, like E2F target genes, and the senescence associated secretory phenotype (IL-6, IL-8, PAI-1) can also induce and maintain senescence driven by RAS over-expression (Dimauro and David 2010).
BRAF
BRAF belongs to the serine threonine protein kinase family and acts downstream of oncogenic RAS by activating the MEK-ERK cascade in tumors. BRAF is well studied in the context of melanoma as 70 % of the cases carry activated BRAF mutations. BRAF can be activated by a single amino acid substitution. The glutamic acid to valine substitution (BRAFV600E) is the most commonly occurring, as it is present in 90 % of the cancers involving this gene (Dhomen and Marais 2007). Study of BRAF mutations in Nevi is an interesting model of oncogene-induced senescence. Nevi are benign tumors of melanocytes in which BRAF mutations occur at a very high frequency. Activated BRAF favors the establishment of senescence, rather than its progression towards melanoma. Recent studies demonstrate that BRAFV600 over-expression in melanocytes contributes to an initial proliferative burst of melanocytes followed by a growth inhibitory response associated with a stable proliferation arrest, an increase in p16INK4a and expression of senescence associated SA-β-gal activity (Michaloglou et al. 2005). As shown in RAS, BRAF needs to cooperate with other activated oncogenes or loss of tumor suppressors to escape senescence effect. A recent paper demonstrates that BRAF-induced senescence depends on the secreted protein IGFBP7 and that loss of this protein is a critical step for the development of melanoma (Wajapeyee et al. 2008). BRAFV600E also cooperates with loss of Pten to promote tumor progression and metastatic melanoma (Dankort et al. 2009).
8.1.2.4 Loss of Tumor Suppressor Genes and Senescence
Similar to over expression of oncogenes, tumor suppressor genes loss can also trigger senescence in vitro and in vivo. Senescence induced by loss of tumor suppressor genes (TSG) was first reported for the tumor suppressors PTEN and NF-1. Complete loss of PTEN induces a senescence response termed PTEN loss induced cellular senescence (PICS) that is triggered by p53 activation (Alimonti et al., 2010 b). Both mTOR-induced p53 translation and ARF mediated p53 stabilization cooperate to establish PICS. Pten loss in the mouse prostate promotes the formation of a benign prostate tumor lesion. However loss of p53 in Pten -/- cells and mouse prostate epithelium by-passes PICS, triggering prostate tumor invasiveness (Chen et al. 2005). Loss–of-function or mutations in NF-1, the gene involved in type-I neurofibromatosis, is also associated to senescence both in vitro and in vivo. Neurofibromatosis is a tumor disorder characterized by the development of benign tumor lesions along the nervous system. Histological analyses have demonstrated that dermal neurofibromas are SA-β-gal and p16INK4a positive (Courtois-Cox et al. 2006). NF-1 converts RAS to its inactive GDP form, and reduced levels of NF-1 expression inhibit RAS activity by shutting down its downstream signaling resulting in profound senescence effects (Bardeesy and Sharpless 2006; Courtois-Cox et al. 2006). VHL (von Hippel-Lindau tumor suppressor gene) is the most commonly mutated TSG in renal cell carcinomas and haemangioblastomas in humans. Acute inactivation of VHL in the mouse causes the formation of benign renal tumor lesions characterized by a senescence phenotype. VHL loss induces senescence by up regulating both Rb and p27 in a p53 and Hif-dependent manner. VHL-associated neoplasias need to overcome loss of senescence induced by VHL to become aggressive (Young et al. 2008). RB1 loss in thyroid cells induces the formation of benign adenomas characterized by increased levels of senescence markers. RB1 loss promotes elevated NRAS activity that in turn induces a DNA damage response and p130-dependent cellular senescence (Shamma et al. 2009). This effect is mediated by E2F activation. On this line, a transgenic mouse model over-expressing E2F3 protein in the intermediate lobe of the pituitary gland developed hyperplasia and senescence in the mouse pituitary gland (Lazzerini et al. 2006) (Fig. 8.2).
8.1.3 Autocrine and Paracrine Senescence
Recent findings demonstrate that senescent tumor cells secrete a variety of immune modulators and inflammatory cytokines, referred to as the senescence-associated secretory phenotype (SASP) or senescence secretome, that mediates contradictory effects. The SASP can stimulate the innate and adaptive anti-tumor immune response (a process designated as “senescence surveillance”) leading to tumor clearance (Xue et al. 2007; Kang et al. 2011) but can also promote tumorigenesis by supporting the proliferation of neighboring tumor cells (Rodier et al. 2007; Coppe et al. 2010; Davalos et al. 2010) or by hindering chemotherapy efficacy (Jackson et al. 2012). Oncogene induced senescence is tightly regulated by the SASP. Recent evidences in OIS demonstrate that senescence entry and maintenance depends on the functional integrity of the chemokine receptor 2 (CXCR2), receptor of interleukine 8 (IL8). Interestingly, inactivation of this receptor blocks both replicative senescence and OIS. Indeed cells undergoing OIS secrete multiple CXCR2-binding chemokines that acts in an autocrine manner to reinforce senescence. The release of these chemokines is regulated by NF-kappaB and C/EBPβ transcription factors and coordinately induces CXCR2 expression (Acosta et al. 2008). Another paper demonstrated that IL6 is also required for the execution of OIS, in a cell-autonomous mode. IL6 depletion caused the SASP to collapse and abolished senescence entry and maintenance (Kuilman et al. 2008). Interestingly the transcription factor C/EBPβ cooperates with IL6 to amplify the activation of the inflammatory network, including IL8.
Interestingly, senescent cells can also induce senescence in normal cells acting in a paracrine manner, through the secretion of cytokines and secreted factors in the tumor microenvironment. Therefore senescence can be propagated in tumors and normal tissues in a non cell autonomous manner. Among other factors that can induce paracrine senescence, TGF-β family ligands, VEGF, CCL2 and CCL20 were identified as major regulators. Importantly, IL1α can control paracrine senescence by modulating the SASP of OIS. Indeed IL1α expression can reproduce SASP activation in normal cells, resulting in senescence (Acosta et al. 2013). Therefore, paracrine senescence acts by reinforcing the arrest of cells undergoing OIS. Inactivation of IGFB7, a gene that encodes for a secreted protein, can also control BRAFV600E induced cellular senescence acting in a paracrine manner. Interestingly IGFB7 blocks the activation of the MAPK induced by BRAFV600E over-expression, promoting cell cycle arrest. Therefore IGFB7 acts as a paracrine factor that can reinforce senescence after the initial proliferative outburst induced by BRAF (Wajapeyee et al. 2008).
8.1.4 Autophagy and Senescence
Autophagy is a genetically regulated program responsible for balancing sources of energy in many physiological conditions. This process plays an essential role in removing misfolded or aggregated proteins, clearing damaged organelles, and eliminating intracellular pathogens. Autophagy is generally considered to be a survival mechanism, nevertheless cellular stress can also activate this process, and recent findings suggest that autophagy may be considered an important tumor suppression mechanism. Young and colleagues lately depicted a new role for autophagy as an effector mechanism of senescence. Indeed, autophagy was upregulated upon OIS induction in human fibroblasts, as indicated by accumulation of autophagosomes and increased protein degradation, and was shown to mediate senescence establishment in vitro. These results were further confirmed in murine papillomas in vivo, suggesting that autophagy may play a critical part in tumor-suppression by supporting the role of senescence as a barrier against tumorigenesis (Young et al. 2009). Interestingly, Dorr and colleagues recently showed that therapy-induced senescence mediates a metabolic reprogramming of cancer cells and a consequent strong proteotoxic stress. Interestingly, cancer senescent cells rely on autophagy as a source of energy to counteract this cellular stress and to fuel the senescence response. Such liability of tumor cells on autophagy may be used to selectively eliminate senescent cancer cells in vivo, thus rendering autophagy therapeutically targetable in tumors (Dorr et al. 2013).
8.1.5 Oncogene Induced Senescence and Immune Response
The role of cellular senescence as a barrier to tumorigenesis both in vitro and in vivo has been deeply investigated. Nevertheless, recent findings indicate that tumour regression associated with senescence response also involves non cell-autonomous processes. Xue and colleagues recently showed that p53 reactivation in a liver cancer mouse model expressing oncogenic Nras (NrasG12V), leads to a strong tumour involution associated with senescence induction and with an inflammatory reaction that involves cell subsets from the innate immune response, such as neutrophils, macrophages and natural killer cells. In this model, p53 activation and senescence provoke an up-regulation of cytokines known to recruit and activate cells from the immune response (Xue et al. 2007). These evidences were further supported by a follow up manuscript from Zender and colleagues, indicating that antigen-specific CD4+ T lymphocytes infiltrating the tumour indirectly mediate the clearance of senescent cells in liver cancer by orchestrating monocytes and macrophages activation (Kang et al. 2011). All together these findings support a model made of a cascade of events, designated as “senescence surveillance”, in which cell senescence triggers the recruitment of immune cell subsets and consequently primes an inflammatory response that flows into the clearance of senescent tumours cells. Importantly, the senescence surveillance cannot be ascribed to a single immune cell subset, but it seems to rather depend on the cooperation of distinct cell types (Xue et al. 2007; Kang et al. 2011; Iannello et al. 2013). Multiple findings indicate that cell senescence may also be induced and regulated by non cell-autonomous mechanisms. In line with this evidences, Braumuller and colleagues recently showed that tumour-infiltrating T-helper 1 lymphocytes can trigger senescence in p53 null pancreatic tumors (RIP-Tag2 mouse model). Senescence induction in this model was antigen restricted and was mediated by IFN-γ and TNF-α release from Tag-specific T cells. Interestingly, the permanent growth arrest of β-cells was strictly depending on TNFR1 signaling, and was fully abrogated in absence of p16INKa and p19ARF expression. Notably, IFN-γ and TNFα was not restricted to Tag-expressing β-cancers as this mechanism of action provoke senescence induction in several mouse and human cell lines in vitro (Braumuller et al. 2013). All together these findings add new insights on the interaction existing between tumor-infiltrating immune cell subsets and senescent cells in cancer and may flow into new immunotherapic strategies for the treatment of human cancer.
8.2 PTEN-Loss Induced Cellular Senescence
8.2.1 PTEN Function
PTEN was originally discovered as the tumor suppressor gene frequently lost on chromosome 10q23 (Li et al. 1997). Subsequent studies demonstrated that loss of PTEN function resulted from several genetic mechanisms including small-scale PTEN gene mutations (point mutations, insertions, small deletions), allelic loss at chromosome 10 and epigenetic silencing via hyper-methylation of the PTEN promoter region (Salmena et al. 2008). The relevance of PTEN in cancer has been addressed through the generation of germline knockout Pten mice by several independent laboratories (Di Cristofano et al. 1998; Suzuki et al. 1998). These studies revealed the requirement of Pten for embryonic development. Importantly, heterozygous loss of this tumor suppressor gene in the mouse resulted in the development of cancer of multiple origins as well as in a lethal lymphoproliferative disease (Di Cristofano et al. 1998). In humans, germline loss and mutation of PTEN is observed in a group of autosomal dominant syndromes (PTEN hamartoma tumor syndromes or PHTS) which are characterized by neurological disorders, multiple hamartomas and cancer susceptibility (Hobert and Eng 2009). The most common PHTS associated to PTEN mutations is Cowden Syndrome, followed by Bannayan–Riley–Ruvalcaba syndrome and Lhermitte–Duclos disease. Studies from the mouse have recapitulated a fraction of the features observed in PHTS patients; however, the cooperative genetic or environmental factors contributing to the full symptomatic spectrum in this group of syndromes remain to be defined. PTEN functions as a lipid phosphatase, dephosphorylating the 3′ position of phosphoinoisitde 3,4,5-triphosphate (PIP3). This lipid second messenger is the product of a potent proto-oncogenic kinase, Phosphoinositide 3-Kinase (PI3K), and the trigger for activation of the PI3K pathway (Salmena et al. 2008). The relevance of the PI3K pathway in cancer is highlighted by the elevated number of components within the cascade, whose level or activity is found altered, and represents one of the main targets for cancer therapy (Wong et al. 2010). PTEN can affect tumorigenesis in different modalities. We and others have previously demonstrated that PTEN is a bona fide haploinsufficient tumor suppressor gene and that even subtle variations of PTEN levels can initiate tumorigenesis (Alimonti et al. 2010a). These studies demonstrate that PTEN dosage variations rather than PTEN deletions or mutations account for tumor initiation in different models. On this line, recent evidence demonstrate that PTEN expression is regulated by a complex network of miRNAs and that deregulation of this miRNA network affect PTEN levels promoting tumor initiation (Poliseno et al. 2010). Importantly, despite the main role of PTEN as a negative regulator of the PI3K pathway, recent studies report a number of tumor suppressive activities for PTEN that are exerted from within the nucleus, where catalysis of PIP3 does not appear to represent a central function of this enzyme. PTEN phospatase activity can be inhibited by proteins that interact with PTEN such as P-REX2a that are frequently up-regulated in a variety of tumors (Fine et al. 2009). These PTEN interactors can affect the PI3K signaling by increasing the cellular levels of PI3P through inhibition of PTEN in tumors with low frequency of PTEN mutations or deletions. Recent evidences also demonstrate that PTEN has a phosphatase independent function in the nucleus. PTEN mono-ubiquitination mediates its nuclear import and binding to the APC/CDH1 complex arresting cellular proliferation (Trotman et al. 2007). PTEN also binds and stabilizes the centromere in a phosphatase independent manner and PTEN loss drive DNA damage accumulation in immortalized cells (Shen et al. 2007). Finally PTEN complete loss drives a cellular senescence response termed PICS, which depends on the functional activation of mTOR and that can be targeted for cancer therapy (Alimonti et al. 2010b). Taken together all these evidence demonstrate that PTEN affects tumorigenesis at multiple levels and with several modalities.
8.2.2 Loss of PTEN and Senescence
The tumor suppressor activity of Pten leads to a novel paradigm, which is called ‘obligate haplo-insufficiency’. Although heterozygous loss of PTEN initiates tumorigenesis, complete loss of Pten activates a p53-dependent cellular senescence response, which acts as a fail-safe mechanism by blocking tumor progression. This senescence response has been named PICS, for PTEN loss induced cellular senescence. As recently shown, PICS depends on the functional activation of p53. Indeed loss of Pten doesn’t initiate senescence when p53 is mutated, leading to tumor formation and invasiveness (Berger et al. 2011). In characterizing the mechanisms and features of PICS it has been discovered that PICS is a distinct form of cellular senescence with several unique differences from oncogene-induced senescence (OIS) and replicative senescence. Indeed, PICS can occur in arrested cells treated with aphidicolin, a compound which blocks S phase entry and prevents DNA replication. On the contrary, in OIS, aphidicolin treatment results in the abrogation of senescence. Furthermore, pharmacological and genetic inactivation of ATM has no effect on PICS induction. Similar to OIS, p53 has an important role in PICS. However, in this context p53 upregulation is mainly promoted by mTOR-mediated translation88. Importantly these findings have been also validated in human cancer cell lines in different studies (Kim et al. 2007). In addition, inactivation of ARF does not dramatically alter p53 levels and senescence in vivo, demonstrating the limited role of ARF for PICS. Finally PTEN loss promotes the upregulation of INK4A through the regulation of ETS2 (Ohtani et al. 2001). Thus, PTEN loss drives senescence in two different manners: through p53 upregulation resulting from mTOR hyperactivation, and through INK4A upregulation resulting from disassembly of the CDH1-containing anaphase-promoting complex (APC/C (also known as the cyclosome)–CDH1) and subsequent accumulation of ETS2. Although it is clear that PICS has a central role in blocking tumour progression in prostate tumorigenesis, the extent to which PICS might restrict the development of other tumors is not clear (Figs. 8.2).
8.2.3 Potential Benefits of PICS
Induction of PICS from a therapeutic prospective offers several advantages. Since PICS does not require hyperproliferation and DDR, treatments that promote PICS in cancer cells will promote a stable cell cycle arrest in absence of genomic instability, avoiding the risk of secondary mutations. Moreover PICS can also occur in non proliferating cells and this property may also be used to induce senescence in quiescent cancer-initiating cells (CICs), that contribute to the maintenance of the tumor in many cancer types. As discussed above, PICS depends on the functional activation of mTOR-mediated p53 translation rather than to ARF mediated p53 stabilization, therefore treatments that stabilize p53 act as pro-senescence compounds in PTEN null cells. On the contrary, OIS depends on ARF-MDM2 p53 stabilization to be executed (Palmero et al. 1998). As a consequence, in contrast to OIS, the treatment of Pten null cells with MDM2 inhibitors can greatly increase p53 levels. Thus, PICS represents a promising therapeutic option. Moreover it has been demonstrated that PICS can be evoked even in PTEN null tumors that have lost p53. Indeed a novel SKP2 inhibitor an skp2 inhibitor can efficiently promote senescence in PTEN null; p53 null human cancer cell lines and arrest tumorigenesis without enhancing senescence in normal PTEN wt cells. SKP2 inhibitors act by enhancing p27 levels, another essential player of senescence in tumors. These findings also demonstrate that PTEN deficient tumor cells are more susceptible that normal cells to SKP2 inhibitors and that PTEN deficient cells conserve the capability to undergo senescence even in presence of p53 mutations or deletions (Nardella et al. 2011).
8.3 Pro-senescence Therapy in Tumors: Pre-clinical and Clinical Development
8.3.1 Therapy-Induced Cellular Senescence
Therapy-induced cellular senescence (TIS) occurs in tumors in response to radiotherapy or selected chemotherapy agents and is a potential strategy for cancer treatment (Roninson 2003; Ewald et al. 2010). The mechanisms that mediate TIS in cancer cells are not well defined but are generally linked to DNA damage enhancement. Tumor cells can be forced into senescence by agents of clinical interest in the management of human cancers, such as docetaxel, bleomicina, cyclophosphamide doxorubicin, etoposide and cisplatin. Ionizing radiation can also induce senescence in different cancer cell lines (Chang et al. 1999; Roninson 2002; Gewirtz 2008). Interestingly, many evidences indicate that therapy-induced senescence may contribute to treatment outcome in vivo. Primary murine lymphomas have been shown to respond to cyclophosphamide therapy by engaging a senescence program controlled by p53 and p16INK4a (Schmitt et al. 2002). In addition to this, analysis of senescence markers in human biopsies from cancer patients after neoadjuvant chemotherapy revealed the occurrence of chemotherapy-induced senescence and its association to treatment outcome (te Poele et al. 2002; Roberson et al. 2005; Coppe et al. 2008). Therapeutically advantageous outcome might be achieved through the combination of TIS with treatments aimed to eliminate cancer senescent cells. On this line, recent findings indicate that senescence cells can be selectively targeted in murine cancers. Virotherapy has been lately explored to eliminate senescent cancer cells by introduction of oncolytic measles vaccine virus (MeV) (Weiland et al. 2014). In addition, Dorr and colleagues recently showed that senescence cells exhibit specific metabolic requirements that can be pharmacologically targeted to selectively eliminate these cells in vivo. This approach has been shown to improve treatment outcome in a model of TIS (Dorr et al. 2013).
8.3.2 p53 Targeting
Inactivation of p53 functions is an almost universal feature of human cancer cells (Lane et al., 2010b). However several cancers such as prostate cancer or glioblastoma conserve an intact p53 response even at advanced or metastatic stage (Lane et al., 2010b). The tumour-suppressive function of p53 predominantly relies on its function as a transcription factor and it can either positively or negatively regulate the expression of numerous target genes. Experimental models demonstrate that reactivation of p53 in established p53 null tumors leads to regression of lymphomas and sarcoma without affecting normal cells. The mechanism responsible for tumor regression depends on the tumour type, with the main consequence of p53 restoration being apoptosis in lymphomas and cellular senescence in sarcomas (Ventura et al. 2007). Another paper demonstrates that reactivation of p53 in established liver cancer results in tumor regression driven by senescence and activation of an anti tumor immune response (Xue et al. 2007). These and previous studies have inspired therapeutic approaches based on p53 reactivation. The most common approach to reactivate p53 in tumors is the p53 gene therapy. This approach is based on adenoviral delivery of p53 in tumor cell lacking p53. p53 gene therapy is now in widespread use in China (Shi and Zheng 2009), but it has not been approved in USA. Another strategy is activating the p53 through siRNA and antisense RNA’s that block the function of the negative regulators Mdm2, MdmX, and HPV E6 (Jiang and Milner 2002; Zhang et al. 2005; Yu et al. 2006). However these approaches are not yet in clinical trial. The most promising approach to enhance p53 function in tumor cells, currently in the clinic, is the use of small molecules that activate p53 either directly or indirectly. There are several phase I clinical trials open with novel p53-mdm2 interaction inhibitors such as DS-3032b, SAR405838 and the Nutlins family members RO5503781, RO6839921, RO683992 (Table 8.1). These compounds show an increased tolerability for cancer patients compared to previous MDM2-P53 inhibitors that have showed excessive toxicity such as JNJ-26854165 (Wade et al. 2013). The compounds CP-31398 and APR-246 (PRIMA-1 analogue) (Peng et al. 2013) have been shown to enhance apoptosis by reactivating p53 function in cells with mutant p53. This approach has been developed to selectively treat tumors with mutant p53. APR-246 is now in clinical trial in combination with carboplatin in ovarian cancer (Table 8.1). Additional approach to reactivate p53 in cancer cells is targeting SirT1. This protein negatively regulates p53 by promoting deacetylation of p53 and its destabilization (reviewed in van Leeuwen and Lain (van Leeuwen and Lain 2009)). Inhibition of SirT1 can slow down tumor growth activating p53 function. There are several SirT1 inhibitor in pre-clnical trials such as e.g., sirtinol, suramin, tenovins, and 3, 2′, 3′, 4′-tetrahydroxychalcone) that can be administered in single, whereas others such as EX-527 and cambinol need concomitant addition of DNA damaging agents such as etoposide to have an effect on p53 (Lane et al., 2010b). Finally treatment with Src and c-Kit kinase inhibitor Dasatinib also resulted in enhanced p53 activity in human acute myeloid leukemia stem cells (Dos Santos et al. 2013). Further trials are needed to validate whether p53 and senescence are selectively enhanced in tumors treated with p53-targeting compounds.
Table 8.1
Pro-senescence cancer therapeutics in pre-clinical and clinical development
Function | Compound | Stage of development | Refs |
|---|---|---|---|
Mutant p53 reactivation | CP-31398 | Phase I | NCT00900614 |
Sir T1 inhibitors | Sirtinol | Preclinical | Ref. (Lane et al. 2010) |
Suramin, Tenovins 3,2′,3′,4′-tetrahydroxychalcone | |||
P53-MDM2 inhibitors
Related posts: Introduction to Cell Stress Responses in Cancer: The Big Picture Introduction to Cell Stress Responses in Cancer: The Big Picture
 Targeting Hypoxic Adaptations of Cancer Cells: Molecular Mechanisms and Therapeutic Opportunities Targeting Hypoxic Adaptations of Cancer Cells: Molecular Mechanisms and Therapeutic Opportunities
 Inflammatory Dysregulation and Cancer: From Molecular Mechanisms to Therapeutic Opportunities Inflammatory Dysregulation and Cancer: From Molecular Mechanisms to Therapeutic Opportunities
 Cell Cycle Checkpoint and DNA Damage Response Defects as Anticancer Targets: From Molecular Mechanisms to Therapeutic Opportunities Cell Cycle Checkpoint and DNA Damage Response Defects as Anticancer Targets: From Molecular Mechanisms to Therapeutic Opportunities
 The Hsp70 Family of Heat Shock Proteins in Tumorigenesis: From Molecular Mechanisms to Therapeutic Opportunities The Hsp70 Family of Heat Shock Proteins in Tumorigenesis: From Molecular Mechanisms to Therapeutic Opportunities
 Cell-Nonautonomous ER Stress-Mediated Dysregulation of Immunity by Cancer Cells Cell-Nonautonomous ER Stress-Mediated Dysregulation of Immunity by Cancer Cells
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

|