Chapter 16 Primary Immunodeficiencies
• Primary immunodeficiency diseases result from intrinsic defects in cells and mediators of the innate and adaptive immune system.
• Defects in B cell function result in recurrent pyogenic infections. Defective antibody responses are due to failure of B cell function, as occurs in X-linked agammaglobulinemia, or failure of proper T cell signals to B cells, as occurs in hyper-IgM (HIgM) syndrome and common variable immunodeficiency (CVID).
• Defects in T cell function due to ineffective antigen presentation or immune recognition result in susceptibility to opportunistic infections. Other abnormalities of T cells may also lead to immune dysregulation with autoimmunity or overactive immune responses.
• Hereditary complement component defects cause a number of clinical syndromes; the most common affects C1 inhibitor, which results in hereditary angioedema (HAE). Deficiencies of the terminal complement components (C5, C6, C7, and C8) and the alternative pathway proteins (factor H, factor I, and properdin) lead to increased susceptibility to infections with N. gonorrheae and N. meningitidis.
• Phagocyte defects, due to reduced numbers or impaired function, can result in overwhelming bacterial and fungal infections. Failure to kill bacteria and persistence of bacterial products in phagocytes leads to abscesses or granulomas, depending on the pathogen.
• Leukocyte adhesion deficiency (LAD) is associated with a persistent leukocytosis because phagocytic cells cannot migrate into the tissues.
• Antibody deficiencies reflect impaired function of B lymphocytes as a result of intrinsic B cell abnormalities or of defects in T lymphocytes that affect activation and terminal maturation of B lymphocytes
• Combined immunodeficiencies are characterized by impaired development and/or function of T lymphocytes, and functional B cell abnormalities
• Phagocytic cell disorders include defects in development and/or function of myeloid cells (granulocytes, macrophages)
• Complement deficiencies are represented by genetically-determined defects of functional or regulatory components of the complement system
• Disorders of immune regulation include diseases characterized by abnormalities in the mechanisms that control autoimmunity, apoptosis, or extinction of immune responses
• Immunodeficiency syndromes represent a heterogeneous group of PIDs in which defects of one or more components of the immune system are associated with extra-immune manifestations.
• patients with antibody deficiencies are highly susceptible to recurrent pyogenic infections sustained by encapsulated bacteria (Haemophilus influenzae, Streptococcus pneumoniae, Staphylococcus aureus);
• combined immunodeficiencies are characterized by broad susceptibility to infections, that includes not only bacteria, but also viruses and opportunistic pathogens (i.e. ubiquitous germs that do not pose significant harm to immunocompetent individuals);
• patients with disorders of neutrophils are prone to bacterial and fungal infections;
• defects of macrophages result in increased susceptibility to mycobacterial disease;
• defects of Toll-like receptors (TLRs), that act as microbial sensors, cause selective susceptibility to specific types of pathogens;
• defects of complement may lead to increased risk of pyogenic infections, but also of autoimmunity, consistent with the role played by complement in removal of immune complexes.
B lymphocyte deficiencies
Congenital agammaglobulinemia results from defects of early B cell development
B lymphocytes develop in the bone marrow from the hematopoietic stem cell (HSC), through various stages of maturation (see Fig. 9.w1) during which time they rearrange their immunogloulin genes to generate the pre-B cell receptor (see Fig. 9.w2)
during which time they rearrange their immunogloulin genes to generate the pre-B cell receptor (see Fig. 9.w2) . Defects in the expression and/or signaling through the pre-BCR cause congenital agammaglobulinemia with lack of circulating B lymphocytes.
. Defects in the expression and/or signaling through the pre-BCR cause congenital agammaglobulinemia with lack of circulating B lymphocytes.
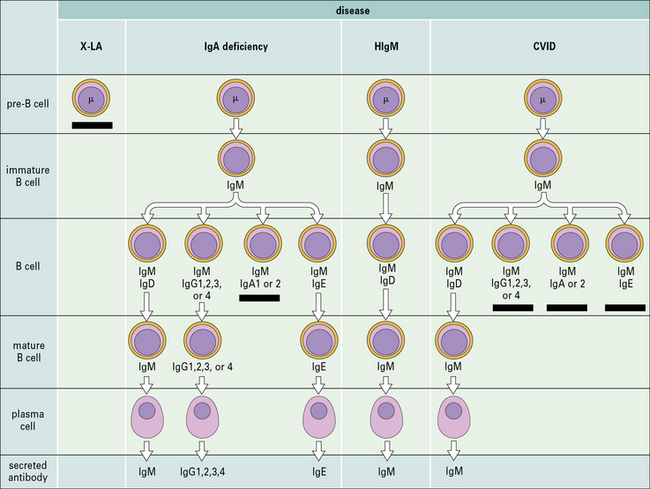
X-linked agammaglobulinemia (XLA) is the prototype of these disorders, and was described by Dr Bruton in 1952. Affected males suffer from recurrent pyogenic infections. They lack serum IgA, IgM, IgD and IgE, and IgG levels are extremely low, usually <100 mg/dL. Circulating B lymphocytes are absent or markedly reduced (<1% of peripheral lymphocytes). Tonsils are absent and lymph nodes are unusually small. XLA is caused by mutations of the Bruton tyrosine kinase (BTK) gene, that encodes an enzyme involved in signaling through the pre-BCR and the BCR (Fig. 16.1). BTK mutations cause an incomplete, but severe, block at the pre-B cell stage in the bone marrow (Fig. 16.2). The BTK protein is also expressed by other cells (including monocytes and megakaryocytes), but its defect does not affect development of these cell types.
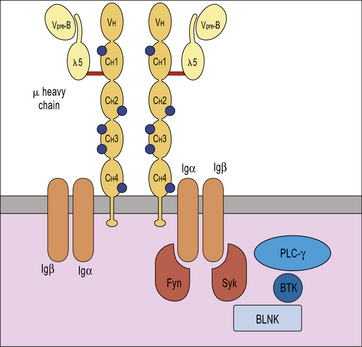
More rarely, congenital agammaglobulinemia is inherited as an autosomal recessive trait, due to mutations of other genes that encode for components of the pre-BCR or of the adaptor molecule BLNK (see Fig. 16.1). In all of these cases, there is a severe block in B-cell development at the pre-B cell stage in the bone marrow. The clinical phenotype is virtually identical to that of XLA.
Defects in terminal differentiation of B cells produces selective antibody deficiencies
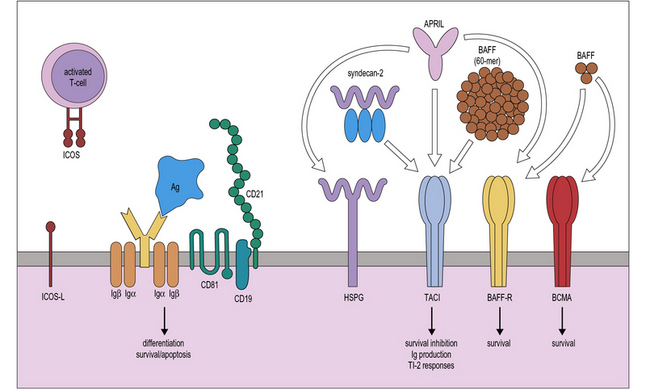
Terminal maturation of B lymphocytes is marked by their differentiation into antibody-secreting plasma cells. Generation of plasma cells is markedly reduced in patients with CVID (see Fig. 16.2), who typically develop progressive hypogammaglobulinemia in the second and third decades of life. CVID is the most common primary immunodeficiency (1:10 000 affected individuals in the general population), characterized by extensive clinical and immunologic heterogeneity. Some patients have a reduced number of circulating B cells, and especially of CD27+ memory B lymphocytes; others show impaired function of T lymphocytes. CVID is usually sporadic, and the underlying molecular defect remains unknown in most cases. However, in some families CVID is inherited as an autosomal dominant or an autosomal recessive trait. A minority of CVID patients carry mutations in genes that play a key role in T-B cell interaction and B cell signaling (Fig. 16.3).
CVID is characterized by reduced levels of specific antibody isotypes
Individuals with CVID have impaired antibody production in response to immunization or to natural infections and there is a virtual absence of plasma cells in lymphoid tissues and in the bone marrow. They suffer from recurrent infections of the respiratory tract (sinusitis, otitis, bronchitis, and pneumonia) sustained by common bacteria (non typeable H. influenzae, S. pneumoniae, etc.); lack of mucosal antibodies results in increased risk of gastrointestinal infection due to Giardia lamblia (Fig. 16.4). They are also highly prone to autoimmmune manifestations (cytopenias, inflammatory bowel disease), granulomatous lesions, lymphoid hyperplasia, and tumors (especially lymphomas). Treatment is based on immunoglobulin replacement therapy and antibiotics. Immunosuppressive and anti-inflammatory drugs may be needed in patients with autoimmune or inflammatory complications.
Defects of class switch recombination (CSR)
Class switch recombination (CSR) is the mechanism by which the μ chain of immunoglobulins is replaced by other heavy chains, resulting in the production of IgG, IgA, and IgE. The process occurs in germinal centres and is accompanied by affinity maturation as described in Chapter 9.
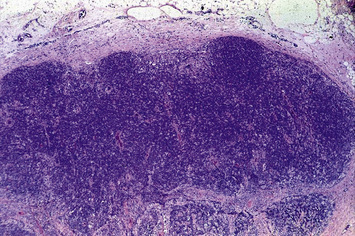
Deficiency of CD40L (X-linked) or more rarely of CD40 (autosomal recessive) results in failure of CSR, with very low or undetectable levels of IgG, IgA, and IgE and normal to increased levels of serum IgM (see Fig. 16.2). In the past, this condition was also known as ‘hyper-IgM syndrome’. In the lymph nodes, primary follicles are present, but germinal centers are absent (Fig. 16.5). Binding of CD40L to CD40 is also important to promote interaction between activated T cells and dendritic cells or monocytes/macrophages. This promotes T cell priming, production of IFNγ and activation of macrophages, that are important in the immune defense against intracellular pathogens. Consistent with this, the clinical phenotype of CD40L and of CD40 deficiency is characterized not only by recurrent bacterial infections, but also by increased risk of early-onset opportunistic infections (Pneumocystis jiroveci pneumonia, cytomegalovirus infection, protracted and watery diarrhoea due to Cryptosporidium). Neutropenia and severe liver disease are frequent. Therefore, CD40L and CD40 deficiency are not pure antibody deficiency, but rather represent examples of combined immunodeficiency. Treatment of these disorders is based on administration of immunoglobulins and antibiotics, but often requires hematopoietic stem cell transplantation (HSCT).
T lymphocyte deficiencies
Severe combined immunodeficiency (SCID) can be caused by many different genetic defects
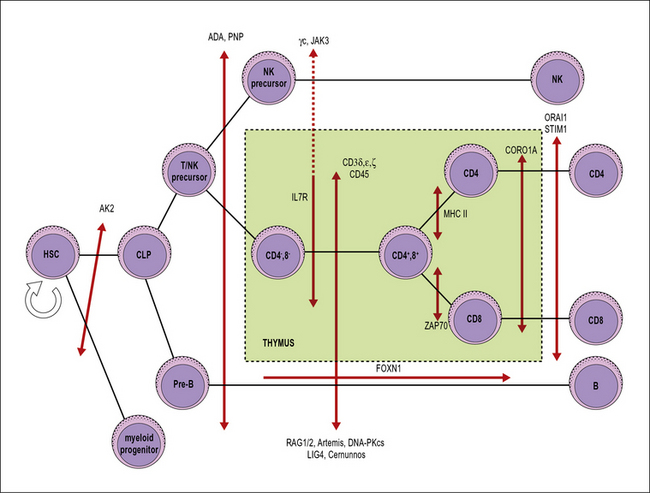
SCID includes a heterogeneous group of genetic disorders that affect various stages of T lymphocyte development or function (Fig. 16.6). The main pathophysiology mechanisms of SCID (and the associated diseases) are:
• impaired survival of thymocytes and T lymphocytes (reticular dysgenesis, adenosine deaminase deficiency, purine nucleoside phosphorylase deficiency);
• defective cytokine-mediated expansion of lymphoid progenitors (X-linked SCID, JAK3 deficiency, interleukin-7 receptor deficiency);
• defective expression of the pre-T cell receptor (deficiency of RAG1, RAG2, and of other components of the V(D)J recombination machinery);
• defective signaling through the pre-T cell receptor (deficiency of CD3 chains, CD45 deficiency);
• impaired positive selection of CD4+ or of CD8+ lymphocytes (HLA class II deficiency and ZAP-70 deficiency, respectively);
• defective egress of T lymphocytes from the thymus (coronin 1A deficiency);
• impairment of calcium flux and of T lymphocyte activation (Stim1, Orai1 deficiencies).