Fig. 6.1
Acute and chronic regulation of aldosterone production. (a) The acute actions of Ang II and extracellular potassium (K+) on adrenal glomerulosa cell aldosterone production. Ang II binds type 1 Ang II receptors (AT1R) activating phospholipase C (PLC) to release diacylglycerol (DAG) and inositol 1,4,5-triphosphate (IP3). IP3 binds the IP3R on the endoplasmic reticulum, releasing calcium and raising cytosolic calcium concentrations. DAG activates protein kinase C (PKC) and protein kinase D (PKD) and phospholipase D (PLD). Ang II also activates PLD in part through PKC. Small increase in extracellular K+ depolarizes the glomerulosa cell, activating voltage-dependent L- and T-type calcium channels, increasing Ca2+ influx. Ca2+/calmodulin-dependent protein kinases (CaMK) and PKD increase StAR and its phosphorylation leading to increased cholesterol movement into the mitochondria. Within the mitochondria, cholesterol is converted to pregnenolone by P-450 side-chain cleavage (CYP11A1) which is then metabolized to aldosterone. (b) The chronic actions of Ang II and K+ on adrenal glomerulosa cell aldosterone production. Ang II binds AT1R to activate PLC activity, which releases DAG and IP3. DAG activates PKC and PKD, and IP3 causes the release of intracellular calcium and the activation of CaMK kinase (CaMKK) and CaMK. Small increases in extracellular K+ also depolarize the glomerulosa cell, increasing calcium influx. PKC activation inhibits the transcription of 17α-hydroxylase (CYP17), while CaMK and PKD increase transcription of aldosterone synthase (CYP11B2). This occurs through increased expression and phosphorylation of specific transcription factors that include NURR1 and CREB. The increase in CYP11B2 raises the capacity to produce aldosterone
ACTH is also able to stimulate aldosterone production acutely both in vivo and in vitro. The adrenal cortex including ZG expresses high levels of the melanocortin receptor 2 (MC2R). ACTH binds to MC2R and activates adenylate cyclase via the heterotrimeric G protein, Gs [16]. Adenylate cyclase produces cyclic AMP (cAMP) which stimulates the activity of cAMP-dependent protein kinase or protein kinase A. PKA induces the expression and phosphorylation of StAR leading to increased cholesterol delivery to the inner mitochondrial membrane [46]. In addition to stimulating cAMP-induced PKA activation, ACTH is able to promote calcium influx likely through PKA-mediated phosphorylation of L-type calcium channels [47, 48], increasing cytosolic Ca2+ concentration and enhancing adenylate cyclase production and aldosterone production [49, 50].
ZG capacity to produce aldosterone is dependent on CYP11B2 expression which apparently results from increased transcription of the gene [51]. Activation of transcription seems to rely on the activation of transcription factors that bind to a cAMP response element (CRE) found in the proximal region of CYP11B2 promotor [52]. In addition, both Ang II and K+ rapidly induce the expression of the nuclear hormone receptor NURR1, which also binds the promotor and activates CYP11B2 transcription [53, 54]. The expression of NURR1 is also increased in adrenal aldosterone-producing tumors and may play a role in the increase in tumor CYP11B2 expression [55].
Primary Aldosteronism
Introduction
Many studies have been conducted to understand the pathophysiology of PA since Dr. Conn’s first description of a patient with APA. After describing the disease, Dr. Conn initially thought that APA could be the cause of up to 20% of patients with arterial hypertension [56]. Subsequently, many physicians undertook a search for adrenal APAs in hypertensive patients and were disappointed when the prevalence of PA appeared to be much lower than expected [57, 58]. This led to the characterization of PA as a rare disease, which at the time was estimated to represent only about 1% of the hypertensive population [59]. However, with the application of aldosterone to renin ratio (ARR) [60] as a screening test for PA, the estimate of the prevalence of PA increased dramatically [61]. The prevalence of PA appears to increase according to the severity of the hypertension [62], and in those patients with resistant hypertension, the prevalence of PA is approximately 20% [8]. It is now recognized that there are several subtypes in PA. APA and IHA are the most common subtypes of PA, and unilateral adrenal hyperplasia, aldosterone-producing adrenocortical carcinoma, FH, and ectopic aldosterone-producing adenoma or carcinoma are known as much less common forms of PA [2]. The currently described PA subtypes and their prevalence are summarized in Table 6.1.
Table 6.1
Subtypes of primary aldosteronism
Subtype | Frequency | Treatment |
|---|---|---|
APA | 30–50% of PA | Adrenalectomy |
IHA | 50–70% of PA | MRA |
Unilateral adrenal hyperplasia | <5% of PA | Adrenalectomy |
Aldosterone-producing adrenocortical carcinoma | <0.5% of PA | Adrenalectomy |
Familial forms | ||
FH-I or GRA | <1% of PA | Dexamethasone or MRA |
FH-II | 5% of PA | Adrenalectomy or MRA |
FH-III | <1% of PA | Bilateral adrenalectomy or MRA |
Familial forms due to CACNA1D or CACNA1H mutations | Extremely rare | MRA, CCB |
Diagnosis
In clinical practice, early detection of PA and targeted treatment for excess aldosterone is essential to prevent cardio- and cerebrovascular complications. According to the Endocrine Society Clinical Practice Guidelines for diagnosis and treatment of PA published in 2016, the diagnosis of PA is a three-step process, comprising (1) case detection, (2) confirmatory testing, and (3) subtype classification [63]. The Endocrine Society Guidelines [63] state the categories of patients with a higher prevalence of PA, who should be screened for the disease:
Sustained blood pressure above 150/100 mmHg on each of three measurements obtained on different days
Hypertension (blood pressure > 140/90 mmHg) resistant to three conventional antihypertensive drugs (including a diuretic)
Controlled blood pressure (<140/90 mmHg) on four or more antihypertensive drugs
Hypertension and spontaneous or diuretic-induced hypokalemia
Hypertension and adrenal incidentaloma
Hypertension and sleep apnea
Hypertension and a family history of early-onset hypertension or a cerebrovascular incident at a young age (<40 years)
Hypertension and a first-degree relative with PA
In addition to those listed above, there are several studies supporting a positive relationship between aldosterone excess and diabetes mellitus and metabolic syndrome. Fallo et al. [64], in a large population of 466 hypertensives, reported a higher prevalence of metabolic syndrome in PA patients compared to essential hypertensives (41.1 vs. 29.6%) and a higher prevalence of hyperglycemia (27.0% vs. 15.2%). Moreover, diabetes mellitus was found to be more prevalent in patients with PA than in 338 matched controls (23 vs. 10%) in the German Conn’s registry population [65]. Considering these findings, hypertensive patients with metabolic syndrome and type 2 diabetes with resistant hypertension are also strong candidates to be considered in the screening for PA [66].
The ARR is currently the most reliable means available for screening for PA [63]. However, there are some issues on the measurement and its interpretation. The most significant confounding factor affecting renin and aldosterone measurements is concurrent or recent use of antihypertensive drugs [67]. In particular, current recommendations suggest withdrawing mineralocorticoid receptor (MR) antagonists and potassium-wasting diuretics for 4 weeks before ARR testing [63]. In addition, the patient ideally should not be taking several common antihypertensive medications that include angiotensin-converting enzyme (ACE) inhibitors, angiotensin receptor blockers (ARB), beta-blockers, and dihydropyridine calcium channel blockers (CCBs) for at least 2 weeks prior to the test [68, 69]. The complete cessation of all antihypertensive treatment is usually unnecessary because there are substitute medications such as non-dihydropyridine CCB, verapamil, and alpha-blockers that have a minimal effect of the ARR [63]. In some patients with severe PA, treatment with MR antagonists cannot be safely discontinued; in this setting, PA-related testing can be pursued as long as renin is suppressed [63]. With regarding to the interpretation of ARR, there is no general consensus on ARR cutoff values, and individual centers use different standards and assays, with a subsequent wide variation in the sensitivity and specificity of the test. Another limitation of ARR is that in the presence of very low renin levels, the ARR may be elevated even when plasma aldosterone is also low which is inconsistent with PA [63]. To avoid this problem, some investigators include a plasma aldosterone concentration of >15 ng/dl within screening criteria [2].
The Endocrine Society Guidelines recommend that patients with a positive ARR undergo confirmatory testing. However, the choice of the test remains a matter of debate, and there is not enough evidence to recommend one over the others. Four different confirmatory tests have been included in the Endocrine Society Guidelines [63]: (1) oral sodium loading with urinary aldosterone, (2) saline infusion with plasma aldosterone, (3) fludrocortisone suppression with plasma aldosterone, and (4) captopril challenge with plasma aldosterone and plasma renin activity. Furosemide upright posture with measurement of plasma renin activity is also in used in Japan [68]. A preliminary study by Ahmed et al. [70] suggests that a seated saline infusion test may be superior to standard recumbent saline infusion test in terms of sensitivity for detecting PA, especially posture-responsive forms, and may represent a reliable alternative to fludrocortisone suppression test. Although these confirmatory tests have variable characteristics in terms of sensitivity, specificity, and reliability, the choice of confirmatory test is commonly determined by considering cost, patient compliance, laboratory routine, and local expertise [63].
For patients with biochemically confirmed PA, it is important to differentiate unilateral and bilateral disease in order to direct the patient toward appropriate therapy: unilateral adrenalectomy for APA and medical treatment with MR antagonists for IHA. As an initial study in subtype testing, all patients should undergo adrenal computed tomography (CT) to rule out the rare, but often fatal, adrenocortical carcinoma [63]. In the absence of cancer, all patients for whom unilateral adrenalectomy is feasible and desired by the patient should undergo adrenal vein sampling (AVS). AVS is highly recommended according to a recent meta-analysis [71], demonstrating that imaging studies show poor sensitivity and specificity for defining the source of aldosterone overproduction. However, there are some drawbacks in AVS. AVS is a technically challenging, invasive, and poorly standardized procedure [72]. During AVS, the blood sample is often obtained near the orifice of the vein and is potentially diluted with other blood, introducing an error in the measurement of aldosterone levels. For this reason, it is necessary to measure cortisol concentrations simultaneously to correct for any dilution. Three AVS protocols are discussed in the Endocrine Society Guidelines [63]: (1)sequential or simultaneous unstimulated bilateral AVS, (2)sequential or simultaneous bilateral AVS followed by bolus cosyntropin-stimulation, and (3) continuous cosyntropin infusion with sequential bilateral AVS; however, there is not enough evidence to recommend one of these protocols over the others.
In patients with an onset of confirmed PA earlier than 20 years of age and in those who have a family history of PA or stroke at a young age (<40 years), the Endocrine Society Guidelines recommend genetic testing for FH-1 (glucocorticoid remediable aldosteronism) which is inherited in an autosomal dominant fashion and is responsible for 1% of cases of PA [63]. In very young patients with PA, genetic testing for germline mutations in the KCNJ5 gene is also recommended for FH-III [63].
Treatment
Treatment options for PA depend on the subtype of PA (Table 6.1).
Unilateral PA: Unilateral adrenalectomy is the treatment choice for unilateral PA because patients have resolution of hypokalemia in nearly all cases and improvement or cure of arterial hypertension in up to 60–70% of patients postoperatively. If a patient is unable or unwilling to undergo surgery, medial treatment including an MR antagonist is recommended [63]. Factors that have been reported to predict cure after adrenalectomy are positive preoperative response to spironolactone [73, 74], short duration of hypertension [73, 75–77], family history of hypertension in one or no first-degree relative [77], preoperative use of two or fewer antihypertensive agents [77], higher ARR, and 24-h urinary aldosterone secretion [73, 77].
Bilateral PA: Medical treatment with an MR antagonist is recommended to treat bilateral PA. Currently there are two alternatives in MR antagonists: spironolactone and eplerenone. Spironolactone is the drug most studied for treatment of PA, although it is a nonselective aldosterone antagonist. The starting dose for spironolactone should be 12.5–25 mg/d in a single dose. The lowest effective dose should be found by gradually titrating upward, if necessary, to a maximum dose of 100 mg/day [63]. This medication can have some side effects in men that include erectile dysfunction and gynecomastia. Eplerenone is a newer selective MR antagonist without antiandrogen and progesterone agonist effects [78]. The starting dose for eplerenone is 25 mg twice daily. In patients with stage III chronic kidney disease, clinicians should administer MR antagonists with caution because of the risk of hyperkalemia. Administering MR antagonists in patients with stage IV renal disease should be avoided [63]. Amiloride (Midamor), an epithelial sodium channel antagonist, also has some advantages although less efficacious than spironolactone. Being a potassium-sparing diuretic, amiloride can effectivly treat both hypertension and hypokalemia in patients with PA [63].
Molecular Mechanisms Underlying Sporadic and Familial PA
Over the last five years, the use of next-generation sequencing (NGS) has dramatically increased our understanding of the genetic causes of PA. Taking advantage of these next-generation technologies, substantial efforts have been directed at defining the pathogenic and molecular mechanisms responsible for autonomous aldosterone overproduction in both sporadic and familial forms of PA. The use of NGS has resulted in the identification of several genes underlying hereditary forms of PA and/or APA, such as KCNJ5 [11], ATP1A1, ATB2B3 [12], CACNA1D [13, 14], and CACNA1H [15].
KCNJ5 Mutations
The KCNJ5 gene encodes the inward-rectifying K+ channel GIRK4, a member of the G-protein activated K+ channel subfamily. This potassium channel is expressed at the plasma membrane of different cell types, forming a homotetramer or a heterotetramer with GIRK1 (encoded by KCNJ3) [79]. Mutations in the KCNJ5 gene have been implicated in the pathogenesis of both FH-III and sporadic APAs [11]. In vitro studies demonstrate that the mutations in KCNJ5, which are located near or within the GYG motif of the ion selectivity filter of the channel, result in pathological sodium permeability, cell membrane depolarization, opening of voltage-gated calcium channels, and increases in intracellular calcium concentration, which in turn activate the transcription of CYP11B2 and increase aldosterone production [11, 80, 81] (Fig. 6.2a). FH-III is a rare inherited form of PA [82]. The disorder is transmitted in an autosomal dominant fashion. In most cases, with the exception of the families carrying the p.G151E mutation [83] and the p.Y152C mutation [81], affected members present with early onset and particularly severe forms of PA, with profound hypokalemia and severe resistant hypertension, requiring bilateral adrenalectomy to control blood pressure.
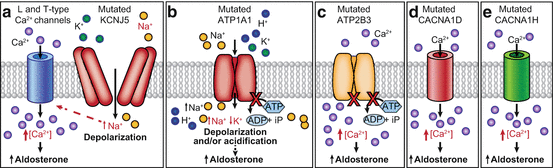
Fig. 6.2
Molecular mechanisms causing autonomous aldosterone production in adrenal cells harboring somatic or germline mutations. (a) The G protein-activated inward rectifier potassium channel GIRK4 which is encoded by the KCNJ5 gene physiologically keeps the cell in a hyperpolarized state. The mutations located near or within the ion selectivity filter of the channel cause a loss of ion selectivity, increased sodium conductance, cell membrane depolarization, and opening of voltage-gated calcium channels, leading to increased intracellular calcium concentration. (b) ATP1A1 gene encodes the α1-subunit of the Na+/K+ ATPase. The Na+/K+ ATPase plays a key role in maintenance of the resting membrane potential and cellular excitability. Mutations in ATP1A1 gene affecting the ion binding capacity disrupt this function leading to cell membrane depolarization and/or cytosolic acidification. (c) Ca2+-ATPase which is encoded by the ATP2B3 gene pumps calcium out of the cell. Loss of this function leads to an increased intracellular calcium concentration. (d) Cav1.3 is the α1-subunit of an L-type voltage-gated calcium channel. Mutations in CACNA1D gene cause activation of the channel at a lower depolarized state, suppress the channel’s inactivation, or directly increase the current flux of calcium. (e) The CACNA1H gene encodes the α1-subunit of the T-type, low voltage-dependent calcium channel Cav3.2. Mutated CACNA1H displays impaired channel inactivation and activation at more hyperpolarized potentials, resulting in increased intracellular calcium levels
Choi et al. [11] originally demonstrated two recurrent KCNJ5 somatic mutations (p.L168R and p.G151R) in 8 out of 22 sporadic APAs. Several centers have investigated the prevalence of KCNJ5 mutations in APAs and have observed variations based on sex and race. In particular, KCNJ5 mutations are much more common in East Asians compared to that seen in Europeans (70–75% vs. 35–40%) [84, 85]. A recent meta-analysis including 13 studies for a total of 1636 APA patients showed an association of KCNJ5 mutations for females as well as for subjects with pronounced hyperaldosteronism, larger tumor size, and younger age at diagnosis [86]. The reasons for this gender dimorphism remain to be elucidated. In vitro studies demonstrated that overexpression of mutant KCNJ5 (p.G151R and p.L168R) in the HAC15 human adrenocortical cells increased CYP11B2 transcript levels as well as CYP11B1 compared with cells expressing wild-type KCNJ5 [80]. A significant increase in transcriptional regulators of CYP11B2, NURR1and NOR-1, was also observed in HAC15 cells expressing the mutant KCNJ5 [80]. Less common somatic mutations which locate near or within the ion selectivity filter of the channel have also been identified [14, 87–94].
The utility of hybrid steroids such as 18-oxocortisol and 18-hydroxycortisol for PA subtype classification has long been studied [95–102]. Recent research has revealed that patients with APA harboring a KCNJ5 mutation are much more likely to have elevated 18-oxocortisol levels compared with other genotypes [102]. In vitro studies also confirmed that a KCNJ5 mutation in HAC15 cells increased 18-oxocortisol production [45, 103]. Therefore there remains potential of hybrid steroid measurement to differentiate IHA from APA in PA patients of East Asian decent where 70% of APA have KCNJ5 somatic mutations [100]. However, in Europeans the use of this particular steroid would be limited because of the lower percentage of patients with KCNJ5 mutations. APAs with KCNJ5 somatic mutations tend to be composed of ZF-like lipid-rich clear cells with high expression of CYP17 and CYP11B1 as well as CYP11B2, while ATP1A1, ATP2B3, and CACNA1D mutated APAs present more frequently ZG-like compact cells with high expression of CYP11B2 [14, 104–106], supporting the findings of increased hybrid steroid production in KCNJ5 mutants.
ATP1A1 and ATP2B3 Mutations
The ATP1A1 gene encodes the α1-subunit of the Na+/K+ ATPase, while the ATP2B3 gene encodes the plasma membrane Ca2+ ATPase isoform 3. Physiologically, the Na+/K+-ATPase exchanges 3 cytoplasmic Na+ ions for 2 extracellular K+ ions for each ATP molecule hydrolyzed, thereby sustaining appropriate ion gradients to allow maintenance of the resting membrane potential. The plasma membrane Ca2+ ATPase isoform 3 transports cytoplasmic calcium ions out of the cell, thus playing a key role in regulating intracellular calcium homeostasis. Somatic mutations in ATP1A1 and ATP2B3 have been identified by Beuschlein et al. [12] in three and two out of nine sporadic APAs, respectively. Similarly, Azizan et al. [14] identified somatic mutations in ATP1A1 in four out of ten sporadic APAs which are composed of ZG-like compact cells. In a large cohort of APAs (n = 474), collected through the European Network for the Study of Adrenal Tumors (ENS@T), somatic mutations in ATP1A1 and ATP2B3 have been reported in 5.3% and 1.7% of APAs, respectively [107], while the prevalence of these somatic mutations seems to be lower in East Asian populations [90].
Cell-based studies demonstrated that ATP1A1 mutations lead to cell membrane depolarization, increased CYP11B2 transcription, and aldosterone production through two different mechanisms: (1) impaired ATPase activity due to a loss of pump function and (2) inward ouabain-sensitive Na+ current or proton-dependent current (Fig. 6.2b) [12, 87]. The intracellular mechanism responsible for the upregulation of CYP11B2 and autonomous aldosterone overproduction in adrenocortical cells carrying an ATP1A1 mutation is believed to be via an increase in intracellular calcium concentration, as occurs in APAs harboring a mutation in KCNJ5. However, a recent study reported no increase in intracellular calcium concentration in H295R adrenocortical cells expressing ATP1A1 mutations [108]. Of note, cytosolic pH measurement revealed an acidification of mutant ATP1A1-expressing cells due to a pathological H+ leak, and the possible contribution of cellular acidification to aldosterone overproduction that was supported by the demonstration that cytosolic acidification with Na+-acetate stimulated CYP11B2 transcription and aldosterone production in untransfected H295R cells [108]. A recent in vitro study showed that a mutation in ATP2B3 (p.L425_V426del) promotes aldosterone production through at least two different mechanisms: (1) a reduced Ca2+ export due to loss of the physiological pump function and (2) an increased Ca2+ influx due to opening of depolarization-activated Ca2+ channels as well as a possible Ca2+ leak through the mutated pump (Fig. 6.2c) [109].
CACNA1D Mutations
Calcium channels mediate the entry of calcium ions into excitable cells and are involved in a variety of cellular processes, including muscle contraction and hormone release. Adrenal glomerulosa cells express both L-type and T-type voltage-gated calcium channels [16]. The CACNA1Dgene encodes the voltage-dependent L-type calcium channel subunit alpha-1D (Cav1.3) that contains four homologous repeats (I-IV). Azizan et al. [14] identified seven different somatic CACNA1D mutations, while Scholl et al. [13] identified four mutations in sporadic APAs without KCNJ5 mutations. Somatic mutations in the CACNA1D gene have been identified in around 8–11% of APAs from Western countries and unlike in KCNJ5, ATP1A1, and ATP2B3, these CACNA1D mutations have been observed throughout the gene affecting all four homologous repeats [13, 14, 107]. Electrophysiological experiments revealed channel activation at less depolarized potentials, leading to increased Ca2+ influx, which is associated with enhanced aldosterone production (Fig. 6.2d) [13].
Related posts:
 Pheochromocytomas and Paragangliomas: Genetics and Pathophysiology
Pheochromocytomas and Paragangliomas: Genetics and Pathophysiology
 Adrenal Zonation and Development
Adrenal Zonation and Development
 Diagnosis and Management of Adrenal Insufficiency
Pheochromocytomas and Paragangliomas: Genetics and Pathophysiology
Diagnosis and Management of Adrenal Insufficiency
Pheochromocytomas and Paragangliomas: Genetics and Pathophysiology
 Adrenal Cushing’s Syndrome: Updates on Overt and Mild Hypercortisolism
Adrenal Cushing’s Syndrome: Updates on Overt and Mild Hypercortisolism
 Diagnosis and Management of Adrenal Insufficiency
Diagnosis and Management of Adrenal Insufficiency
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


