Part 1 / Diabetic Neuropathy
Why would patients who for so long were using only one to two injections of insulin per day who had no access to intensive glucose monitoring, blood pressure (BP) or lipid management have exceeded all expectations by minimizing their prevalence of complications? Other patients with T1DM develop proliferative retinopathy and chronic kidney disease despite remaining vigilant in maintaining their prescribed A1C levels. Clearly, factors other than simple glycemic control factor into long-term outcomes for all patients with diabetes.
more AGEs become receptor bound, the greater the likelihood of developing a complication over time. However, patients, such as the Joslin Gold Medalists, appear to have been provided with a truly unique gift from their parents known as a “soluble” AGE. As the AGE binds to the RAGE, a small protein ligand breaks free of the receptor effectively “blocking” the complication pathway from progressing forward. Patients who can produce the “soluble RAGE” are likely to be symptom free, whereas those patients who lack the soluble component are prone to develop the complications.
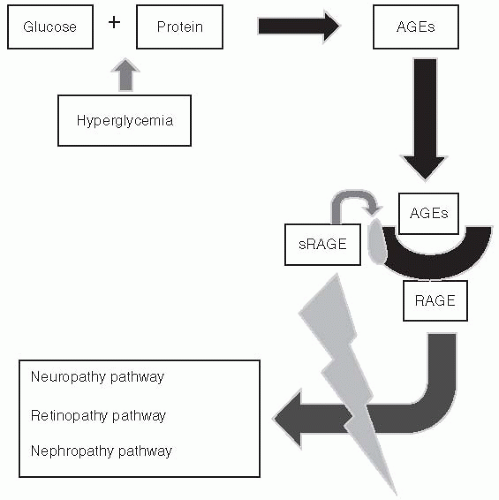 Figure 5-1 • Protective Mechanism of the Advanced Glycation End Product (AGE) Pathway. AGEs form via nonenzymatic glycation of proteins, lipids, and nucleic acids. Once formed, AGEs promote vascular stiffness and alter cellular receptor signaling by binding with AGE receptors (RAGE). The greater the number of AGEs bound to RAGE, the more enhanced oxidative stress becomes resulting in a downstream cascade of events, which, over a number of years, will often result in a number of microvascular complications. However, in patients who “escape” microvascular disease despite having a history of prolonged hyperglycemia and elevated AGEs, binding of AGEs to RAGE induces the release of a soluble receptor ligand known as sRAGE. sRAGE competitively reduces activation of AGE complication pathways, thereby blocking the downstream cascade mechanism which would otherwise induce microvascular complications. Patients who are genetically prone to producing the sRAGE ligand may be spared complications. Those who cannot produce the sRAGE ligand may be at higher risk of developing microvascular disease. (Adapted from Yan SF, Ramasamy R, Schmidt AM. The RAGE axis: a fundamental mechanism signaling danger to the vulnerable vasculature. Circ Res. 2010:106:842-853.) |
declined from 21.7% in 1994 to 20% in 2007. All other risk factor trends have been increasing during this same reporting period.15 Duration of diabetes and exposure to glycemic burden also impact the risk of complications.16
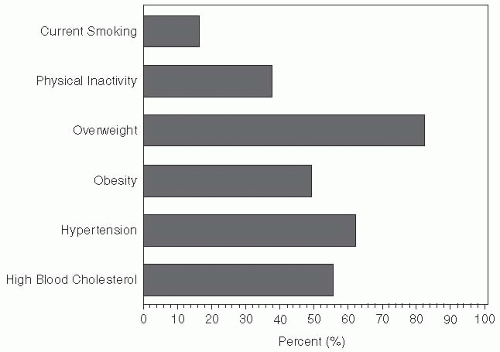 Figure 5-2 • Risk Factors for Complications Among Adults with Diabetes in the United States—2007. According to the CDC, 15.1% of U.S. adults with diabetes smoked, 38.2% reported being physically inactive, 83.5% were overweight or obese, 51.1% were obese based on self-reported height and weight, 67% had hypertension, and 62.6% said that they were diagnosed with high cholesterol. Centers for Disease Control and Prevention Data and Trends. http://www.cdc.gov/diabetes/statistics/comp/fig10.htm. Accessed December 12, 2011. |
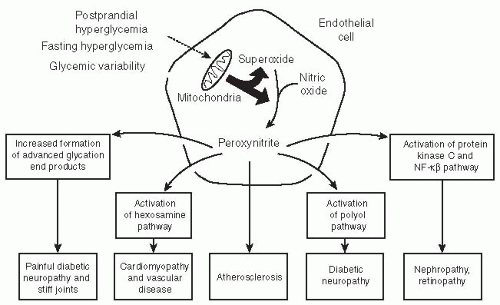 Figure 5-3 • The “Downstream Effects” of Oxidative Stress-Induced Diabetes Complication Pathways. Postprandial and fasting hyperglycemia and glycemic variability result in the production of superoxide within the mitochondria of endothelial cells. NO regulates vascular tone and minimizes adhesion molecule penetration of the vascular walls. When superoxide interacts with peroxynitrate, the endothelial cell’s mitochondrial electron transport system becomes impaired, resulting in endothelial dysfunction. Transcription of endothelial-derived cytokines induces pathways known to activate microvascular complications. Peroxynitrate also favors lipid oxidation leading to atherosclerosis and macrovascular disease. NF-kβ, nuclear factor kappa B. (Adapted from Unger J. Reducing oxidative stress in patients with type 2 diabetes mellitus: a primary care call to action. Insulin. 2008;3:176-184.) |
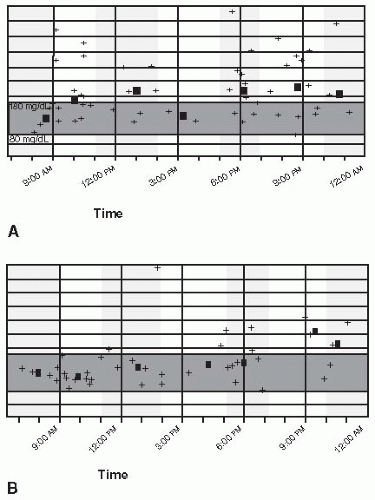 Figure 5-4 • Improvement in Glycemic Variability. A 52-year-old school teacher with stage 4 chronic kidney disease and nonproliferative retinopathy. Despite being on an insulin pump, the patient’s self-blood glucose monitoring suggests wide glycemic variability as shown in panel A. (Each + symbol represents a glucose value obtained during that time over a 2-week interval.) The square represents the average fasting and postprandial glucose values over 2 weeks. After the patient was placed on liraglutide (off label with concurrent use of insulin), his glycemic control and variability were significantly improved. Although this case is suggestive of glycemic variability, MAGE as determined by continuous glucose sensing is the most appropriate model for measuring daily variability as shown in panel B. (Case courtesy of Jeff Unger, MD.) |
circulation in response to tissue ischemia through the release of growth factors and cytokines. The EPCs hone into the ischemic or damaged tissue and stimulate endothelial repair. In addition to traditional cardiovascular risk factors, oxidative stress has been associated with reduction in the number and function of circulating EPCs, whereas an expanded EPC pool decreases cardiovascular mortality.25
 Figure 5-5 • Relationship between Hyperglycemia Markers and Microvascular Complication Risk. Fasting hyperglycemia and postprandial hyperglycemia both contribute to excessive glycation, resulting in a rise in A1C. Acute fluctuations in daily glycemia, as measured by continuous glucose sensors, like A1C will result in a rise in oxidative stress. Complication pathways are activated in response to oxidative stress. Therefore both chronic and acute hyperglycemic abnormalities should be targeted and controlled in order to minimize one’s risk for developing long-term complications. (Adapted from Monnier L, Colette C. Glycemic variability. Should we and can we prevent it? Diabetes Care. 2008;(Suppl 2):S150-S154.) |
are fortunate to experience no diabetes-related complications, whereas those less fortunate with prediabetes may develop retinopathy or painful diabetic neuropathy (Fig. 5-1).
TABLE 5-1. Practical Approaches to Reducing Oxidative Stress | |||||||
|---|---|---|---|---|---|---|---|
|
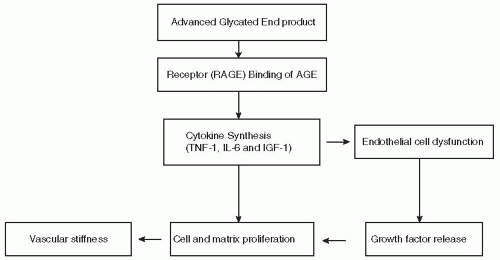 Figure 5-6 • Pathway Linking Advanced Glycation to Diabetes Complications. (See text for explanation of pathway.) |
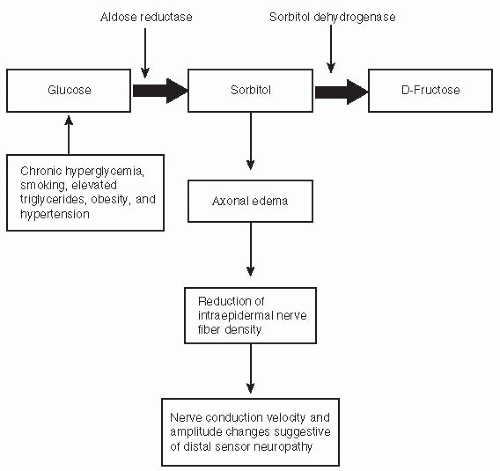 Figure 5-7 • Relationship of the polyol pathway to activation of diabetic neuropathy hyperglycemia results in an increased accumulation of sorbitol and fructose in cells such as neurons and retina tissue, which are unable to eliminate the excess sugars. This changes the osmotic gradient within these susceptible cells, resulting in neuronal dysfunction. |
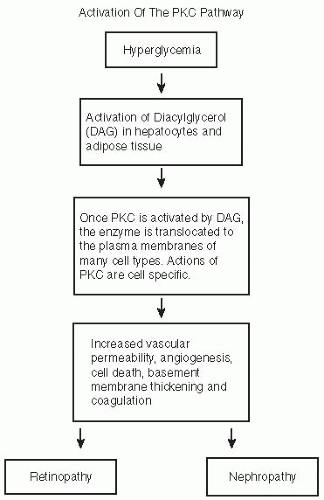 Figure 5-8 • Activation of the PKC Pathway. Chronic hyperglycemia induces the activation of the PKC pathway, which, when combined with increasing oxidative stress and inflammatory cytokine production, results in thickening of the renal arteriolar walls, glomerular basement membrane, and tubular basement membrane. Retinal vessels are also affected by an increase in hyperglycemia-induced PKC signaling. |
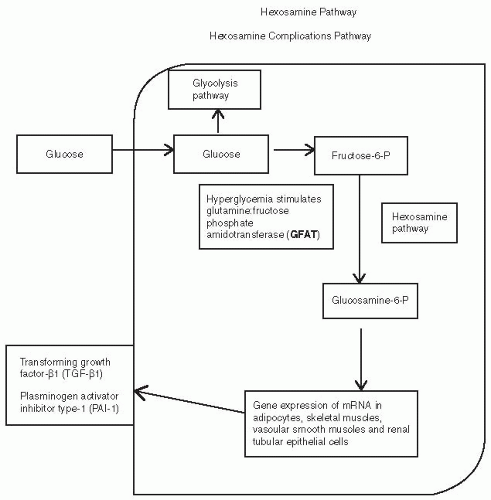 Figure 5-9 • Hexosamine Pathway and Hexosamine Complications Pathway. The normal route of glucose metabolism is through glycolysis. During hyperglycemia, glucose is diverted into the hexosamine pathway in which an enzyme called GFAT (glutamine fructose-6 phosphate amidotransferase) converts the fructose-6 phosphate into glucosamine-6 phosphate. This results in pathologic changes in gene expression within the cell nucleus. Inflammatory cytokines, PAI-1 and TGF-β1 are produced by the cells resulting in vascular disease affecting the kidneys and arteries. (From Schleichler ED, Weigert C. Role of hexosamine biosynthetic pathway in diabetic nephropathy. Kidney Int. 2000;58(Suppl 77):S13-S18.) |
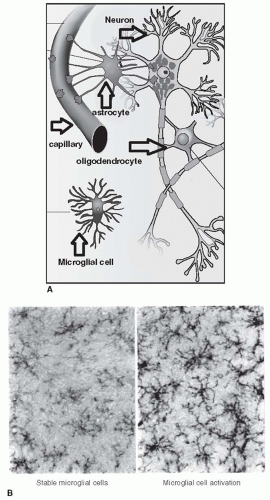 Figure 5-10 • Neuroinflammation. A. Anatomy of the neurovascular unit. B. Microglial cell activation. The neurovascular unit consists of astrocytes, which are modulators of ions and glutamate within neurons and microglial cells. Microglial cells are living sensors, which protect a given neuronal segment. Stable microglial cells (pictured on the left of B) produce a cytokine IL-10, which protect the neuron from inflammation. Microglial cell activation will occur when patients are exposed to physical or emotional stress. Certain medications, such as opioids, will also trigger microglial cell activation (shown on the right of B). Once activated, the microglial cells shift cytokine production to IL-6 and glutamine, both of which are inflammatory and injurious to the neuron. (Adapted from Unger J. Diabetic neuropathy: early clues, effective management. Appl Neurol. 2005:23-30.) |
derangements related to hypertension and hyperlipidemia. DPN is statistically associated with other microvascular complications such as DR and diabetic neuropathy.46
TABLE 5-2. Risk Factors for Diabetic Neuropathy | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
TABLE 5-3. Classification of Diabetic Neuropathy | ||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||
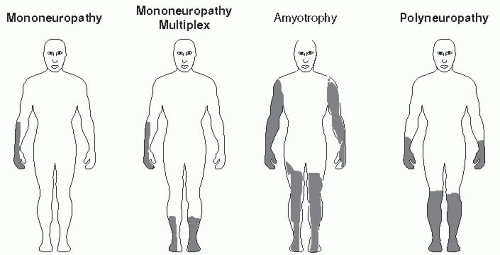 Figure 5-11 • Pain Distribution Associated with Sensorimotor Neuropathies. (Modified from presentation by Charles Argoff at American Conference on Pain Medicine, New York. June 16, 2007.) |
Focal and multifocal neuropathies are confined to the distribution of a single peripheral nerve (mononeuropathy) or multiple peripheral nerves (mononeuropathy multiplex). Mononeuropathies are caused by vasculitis, ischemia of the capillaries supplying the neurons, or nerve infarcts.47 Cranial neuropathy in diabetic patients is rare, typically affecting older persons with a long history of diabetes.48 Cranial nerves III, IV, or VI may be involved (Fig. 5-12). The classic presentation of a cranial neuropathy is acute-onset diplopia with ptosis and papillary sparing associated with ipsilateral headache. Neurologic deficits resolve on average within 21/2 months. Recurrence rates are 25% in patients with diabetes. Advise patients with a cranial neuropathy to wear a patch over the affected eye and to adhere to strategies that improve glycemic control.
Nerve entrapment syndromes begin gradually and may become disabling over time without intervention. Most often, the median, ulnar, peroneal, lateral femoral cutaneous, or tibial nerve within the tarsal tunnel is involved. Entrapment syndromes affect up to 30% of patients with diabetes and should be evaluated carefully in all those with signs and symptoms of neuropathy.49
Carpal tunnel syndrome (median neuropathy) is a clinically relevant problem in 6% of patients with diabetes.50 Painful paresthesias of the fingers may progress to a deep-seated ache, which radiates proximally through the forearm. Symptoms are worse at night. Motor weakness can become progressive, and thenar wasting occurs over time.
 Figure 5-12 • Left VI Nerve Palsy. Left-sided cranial nerve VI palsy in a patient with poorly controlled T1DM (A1C = 9.6%) who recovered spontaneously within 12 weeks after the onset of her symptoms of dyplopia and unilateral headaches. (Photo courtesy of Jeff Unger, MD.) |
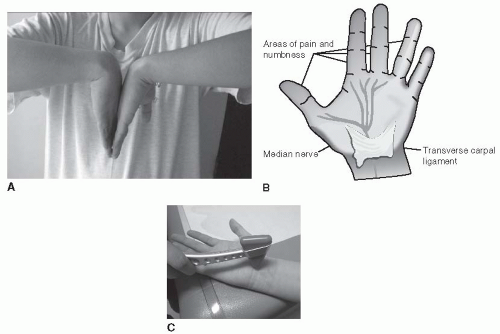 Figure 5-13 • Demonstrates Distribution of Paresthesias in a Patient with a Positive Phalen Test. A. The Phalen test—forearms held vertically and hands held in complete flexion for 1 minute—is positive if paresthesia develops in the median nerve. B. Distribution within 30 seconds. C. The Tinel sign—percussion over the median nerve that induces paresthesia over the distribution of the nerve—is suggestive of carpal tunnel syndrome. |
Ulnar neuropathy occurs in 2% of diabetic patients as a result of nerve compression immediately distal to the ulnar groove beneath the edge of the flexor carpi ulnaris aponeurosis in the cubital tunnel. Alcoholism is a risk factor. Typical symptoms include painful paresthesias in the fourth and fifth digits associated with hypothenar and interosseous muscle wasting. Treatment is conservative. Patients with motor loss and muscle wasting may require surgical intervention.
Compression of the lateral femoral cutaneous nerve (meralgia paresthetica), although uncommon in diabetes, can result in pain, paresthesias, and sensory loss over the lateral aspect of the thigh. Most cases resolve spontaneously. In cases associated with severe pain, allodynia, and disability, corticosteroid injections using focal nerve blocks at the inguinal ligament or surgical decompression may be required.
Tarsal tunnel syndrome is a painful lower limb entrapment syndrome that involves the tibial nerve, as it traverses the tarsal tunnel. The tibial nerve innervates only the muscles of the soles. Walking or standing triggers severe burning pain over the plantar aspect of the foot. A positive Tinel sign on the underside of the medial malleolus with atrophy of the sole muscles are typical
clinical observations. Sensation over the dorsum of the foot is normal. Ankle reflexes are maintained. Nerve conduction studies demonstrate asymmetry compared with the normal leg.
Diabetic truncal radiculoneuropathy affects middle-aged and elderly men. The primary feature is pain of acute onset that resolves spontaneously within 4 to 6 months. The pain— which is worse at night—is described as an aching or burning sensation with superimposed lancinating stabs. Patients describe the location of pain as being in a girdle-like distribution along the lower thoracic or abdominal wall. The pain may be unilateral or bilateral. Patients may experience profound weight loss associated with the onset of their symptoms. Clinical findings range from no abnormalities to sensory loss and painful hyperesthesia in a complete dermatomal pattern.
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


