Fig. 5.1
Adrenal steroid biosynthesis
The mechanism of adrenarche onset remains unknown, although the implication of leptin, IGF1, and ACTH has been conjectured. In addition, MC-2 receptor, CYP19, IGF1, and AR gene polymorphisms have been reported to be associated with PP in some cases.
The physiological role of adrenarche in the pubertal maturational process has never been elucidated.
5.3 Clinical Features
The consequences of exposure to androgen excess in prepubertal girls are:
Development of pubic hair
Development of axillary hair
Acne
Oily skin/hair
Adult body odor
Mood swings and/or behavioral changes
In a recent work, R. Voutilanen reported that 63 girls with PP presented the following characteristics: 89 % adult-type body odor, 70 % oily hair, 56 % acne, 48 % pubic hair, 38 % axillary hair, and 12 % acanthosis nigricans. In 20–50 % of these girls, higher than normal childhood weight was observed. Moreover, a bone age advance of 1–2 years was noted. Although some of these girls presented earlier menarche, normal adult target height was reached [3].
5.4 Evaluation of Girls with PP
The diagnosis of PP is based on the exclusion of other causes of pubertal hyperandrogenism. The focus should be on the age at the onset of signs, the tempo of their changes, and recent acceleration of growth. Examination of the girl should include pubertal staging, a search for other signs of hyperandrogenism, and the evaluation of BMI. Laboratory assessment is limited to the measurement of basal serum 17-OH progesterone, DHEAS, Δ4-androstenedione, and testosterone. An adrenocorticotropic hormone (ACTH) stimulation test must be performed if the basal 17-OH progesterone level is found to be above 2 ng/dL. A bone age evaluation is systematically carried out: in most cases, a 1–2 year advance in bone age is found.
Abdominal MRI should be considered when an adrenal androgen-secreting tumor is suspected.
A definitive algorithm for the evaluation of PP cannot be recommended. We nevertheless propose the following decision tree (REF. Sultan PAG), which has usually been helpful in our clinical experience (Fig. 5.1).
5.5 Differential Diagnosis
PP must be distinguished from a number of pathological conditions:
Precocious puberty
Non-classical congenital adrenal hyperplasia (NC-CAH), due to 21-OH gene mutation
Virilizing adrenal tumors
Exogenous androgen exposure
5.5.1 Precocious Puberty
PP may be a marker for the early onset of puberty: idiopathic central precocious puberty may present with pubic and/or axillary hair [4]. Bone age and growth velocity are accelerated and plasma androgens are abnormally elevated for age (Fig. 5.2d).
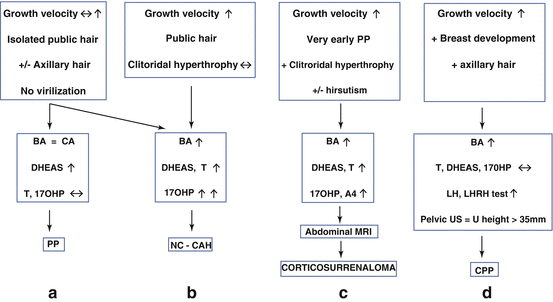
Fig. 5.2
Clinical presentation of pre pubertal hyperandrogenism
5.5.2 Non-classical Congenital Adrenal Hyperplasia (NC-CAH)
NC-CAH is usually related to 21-OH deficiency [5]. In this condition, NC-CAH has an extremely variable presentation. In prepubertal girls, the presenting signs include: PP, cystic acne, accelerated growth, and advanced bone age (Fig. 5.2b).
NC-CAH has been found in 1–30 % of girls with PP. This wide range is likely related to ethnic differences. The biological diagnosis of NC-CAH is suspected when early morning basal 17-OH progesterone is above 200 ng/dL and when an increase in 17-OH progesterone above 1200 ng/dL is found after the ACTH stimulation test [6].
In our experience [7] with a cohort of 40 girls with PP, 24 % of them presented a molecular defect within the 21-OH gene.
5.5.3 Virilizing Adrenal Tumors
Virilizing tumors are rare but not exceptional (Fig. 5.2c): we recently had the opportunity to manage two very young girls referred to our clinic for PP without exaggerated symptoms of virilization. MRI was performed because abdominal palpation revealed an abdominal mass [8]. In both cases, an adrenocortical carcinoma was diagnosed and surgically removed.
Related posts:
 Central Precocious Puberty: From Diagnosis to Treatment
Amenorrhoea and Anorexia Nervosa in Adolescent Girls
Premature Ovarian Failure in Adolescence and Young Adults: From Diagnosis to Therapy and Follow-up for Fertility Preservation
Emergency Contraception
Central Precocious Puberty: From Diagnosis to Treatment
Amenorrhoea and Anorexia Nervosa in Adolescent Girls
Premature Ovarian Failure in Adolescence and Young Adults: From Diagnosis to Therapy and Follow-up for Fertility Preservation
Emergency Contraception
 Delayed Puberty: Impact on Female Fertility
Delayed Puberty: Impact on Female Fertility
 Adolescent Pregnancy and Contraception
Adolescent Pregnancy and Contraception
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


