Keywords
Tumor microenvironment, poxvirus, immunotherapy, immune escape
Introduction
The underlying hypothesis/strategy of our approach to immunotherapy is that by modulating the tumor microenvironment, we will be able to induce an effective systemic tumor-specific response. Our studies utilizing tetramer analysis, functional assessment of tumor-associated, lymph node (LN), and systemic immune populations, and the generation of a family of epitope-specific vaccines have resulted in our defining active immune escape mechanisms in which the tumor microenvironment and draining LN manifest a balance between effector and regulatory cells, resulting in functional anergy in the periphery. We have focused on the use of in situ gene transfer using poxvirus recombinants because we believe this will provide us an opportunity to immunize to the optimal combination of tumor-associated antigens. Our finding that local immune modulation can lead to peripheral responsiveness has led us to hypothesize that local intervention has the potential to enhance the response to tumor antigen-encoding vaccines given in the periphery, thus enhancing systemic effector function. Thus, although there is clear agreement that the development of systemic responsiveness is crucial, if what goes on in the tumor microenvironment, as our studies have shown, blocks the development of effective systemic immunity or, even worse, sets up an environment in which immunization actually hinders the desired response via the expansion of a negative regulatory component as we describe, modulating the tumor–host environment may be critical in inducing effective antitumor immunity. We have translated our findings from our preclinical studies into five clinical trials ( Table 17.1 ) establishing intratumoral gene transfer using poxvirus as safe, allowing prolonged expression of encoded transgenes, and as having the ability to change the immune milieu by altering the infiltrate makeup.
| Vector | Patient Population | Route | Status | Reference |
|---|---|---|---|---|
| Vaccinia WT | Melanoma | Intralesional | Completed | |
| Vaccinia WT | Bladder | Intravesical | Completed | |
| rV-GM-CSF | Melanoma | Intralesional | Completed | |
| rF-GM-CSF | Bladder | Intravesical | In follow-up | |
| PANVAC | Pancreas | Intratumoral | In follow-up |
To be successful, the goal of cancer immunotherapy must be the generation of effective systemic antitumor immunity active at both primary and metastatic sites. Toward this end, tremendous strides have been made in the past decade with a linking of tumor immunotherapy strategies to advances in our understanding of basic immune mechanisms and the development of sophisticated molecular technologies for both tumor and host manipulation. Of particular import has been the elucidation of the cells and mechanisms required for antigen presentation and the regulation of T cell activation. Although the lack of effective immune surveillance and/or successful immunotherapy has in the past been attributed to immune ignorance (lack of recognition) or the induction of antigen-specific tolerance, studies by us and others now definitively show that tumor recognition leads to active immune regulation (reviewed in Dunn et al . and Pure et al . ) via downregulating antigen-presenting cell (APC) function, , inducing T regulatory cell (Treg) responses ; myeloid-derived suppressor cell responses ; and the production of a number of tumor or tumor–stroma-derived factors, such as transforming growth factor-β (TGF-β) and vascular endothelial growth factor (VEGF). Taken together, these mechanisms, all shown to modulate compartments of the antitumor response, have been shown to result in a state of functional anergy. The sum total of such studies has allowed for the identification of regulatory pathways that are rapidly becoming targetable, which in turn has allowed for the development of single- and combined-agent therapeutic strategies. Elegant studies by Schreiber et al . have defined three phases of what is described as immunoediting, which progresses from elimination to equilibrium to escape, highlighting the cells and mechanisms associated with each phase .
The Tumor Microenvironment as Central to the Regulation of Antitumor Responses
Given that the initial immune recognition of tumor takes place in the tumor microenvironment and draining lymph nodes (DLNs), and our and others’ identification that an apparent balance between effector and regulatory populations exist therein, it continues to be our hypothesis that intervening at the tumor site affords the opportunity to enhance local and critically systemic effector function. In addition to the presence of effector/regulatory cells, the tumor microenvironment is the optimal site of tumor antigen. Increasingly, investigators are returning to whole cell vaccines or multiple combinations of characterized antigens for therapy. It is our bias that relying on autologous tumor as a source of antigen, given its expression of multiple antigens identified and not identified, has an optimal chance of inducing a multivalent response. Studies by Sjoblom et al . support this hypothesis by demonstrating an average of 90 mutations per tumor when human breast and colorectal cancers were analyzed , thus identifying a potentially large repertoire of recognizable antigens present in patient tumors. In fact, our focus on the intratumoral approach stemmed from years of experience studying autologous whole cell vaccines . The presence of a source of tumor antigen as well as targetable antigen presentation and regulatory pathways further supports our choice to focus on modulating the tumor microenvironment via local gene transfer as a strategy for optimally immunizing the tumor host.
Tumors Usurp Normal Immune Regulatory Pathways, Resulting in Functional Anergy
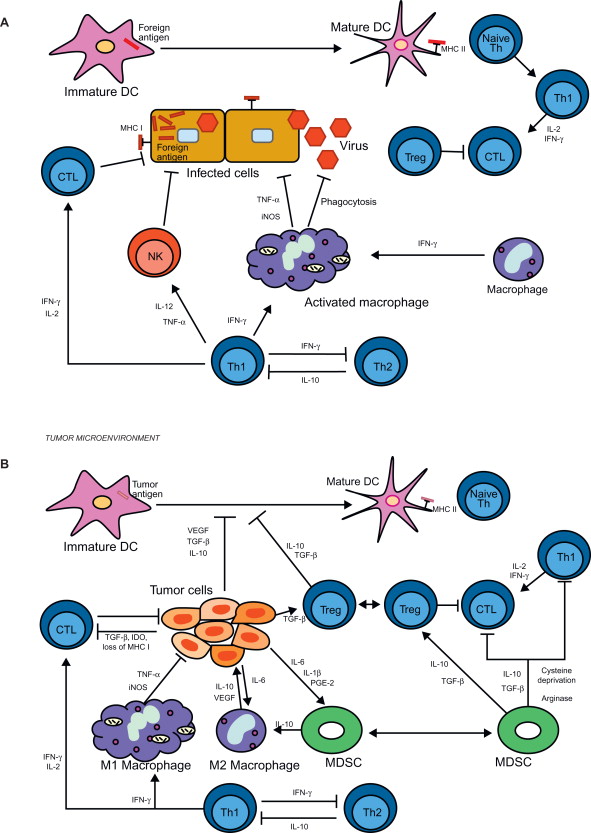
As noted previously, the lack of immune responsiveness to tumor was early on believed to be due to a lack of recognition (ignorance) . An increased understanding of the complexity of the cells/factors involved in the generation and regulation of an immune response, coupled with an increasingly sophisticated armamentarium of reagents and techniques, has led us and others to identify multiple immune mechanisms that are being usurped by tumors with resultant generation of a lack of or reduced responsiveness. Figure 17.1 schematically demonstrates the cells/factors involved in the generation and regulation of T cell responses to nontumor (viral) ( Figure 17.1A ) and the effects of tumor-associated cells/factors on the response ( Figure 17.1B ). Figure 17.1A shows that the immune system is composed of cellular effectors and cytokines that respond to pathogens, and potentially tumor cells, through various mechanisms. APCs, such as dendritic cells (DCs), phagocytize pathogens and present foreign antigen to helper T cells (Th) via the major histocompatibility complex (MHC) class II pathway . This results in the initiation of one of several types of regulatory cytokine-producing Th cell responses, depending on the type of pathogen. In a Th1 response, Th1 cells secrete a set of cytokines, such as interferon-γ, interleukin-2 (IL-2), and tumor necrosis factor-α, that support the proliferation and activation of cytotoxic T lymphocytes (CTLs) and macrophages . Infected cells display foreign antigens on their surface via the MHC class I pathway (MHC I), permitting recognition of these cells by CTLs via the T cell receptor and ultimately cell death . This active T cell response is regulated by Tregs, thus preventing autoimmunity . Figure 17.1B outlines how tumors suppress the formation of an antitumor immune response through the secretion of cytokines and factors into the tumor microenvironment that interfere with the function of various immune effectors. TGF-β, VEGF, and IL-10 interfere with DC maturation, thus preventing effective antigen presentation to T lymphocytes TGF-β also directly interferes with the function of CTLs and promotes the development of Tregs . Overproduction of the enzyme indoleamine 2,3-dioxygenase (IDO) results in depletion of the essential amino acid tryptophan, thus impairing T cell proliferation . The Th1 helper T cell response is also attenuated by IL-10 . Tumors promote the development of myeloid-derived suppressor cells (MDSCs), an immune regulatory population of immature myeloid cells found both in the tumor microenvironment and systemically, through the production of proinflammatory cytokines such as PGE 2 , IL-1β, and IL-6 . MDSCs, in turn, have inhibitory effects on T cells through deprivation of essential amino acids such as cysteine and arginine . MDSCs also promote other immune-suppressive populations such as M2 tumor-associated macrophages and Tregs through production of IL-10, TGF-β, and VEGF . Taken together, these pathways represent a series of overlapping negative regulatory mechanisms, active alone or in combination to inhibit effective antitumor immunity and vaccine therapy.
Poxvirus Strategies for the Induction of Systemic Immunity
Studies in our laboratory, extended to clinical trial ( Table 17.1 ), have taken two complementary approaches toward regulating the immune response to tumor via the manipulation of the tumor microenvironment. In the first, we have utilized poxviral recombinants to infect/transfect tumor and stromal cells in the microenvironment with the cytokine granulocyte–macrophage colony-stimulating factor (GM-CSF), chosen based on its central role for antigen presentation (reviewed previously) and its potent recruitment/activating activities directed at APC. Seminal studies by Dranoff et al . demonstrated that tumors transfected in vitro and used as vaccines induced long-lasting tumor immunity . In this approach, we have developed and tested the ability to use poxvirus to infect/transfect tumor, relying on the tumor to provide the appropriate targetable tumor antigen. In the second approach, informed by our studies demonstrating a potent antigen-specific CTL response ongoing in the tumor microenvironment but not systemically (reviewed here), we have added virally encoded tumor antigen to the therapeutic combination injected into the tumor . As with the first approach, these studies are supported by our preclinical studies showing efficacy and defining mechanism. Recently, we have translated this approach to a phase I clinical trial of endoscopic ultrasound (EUS) targeted intratumoral injection in patients with locally advanced pancreatic cancer.
Use of Recombinant Poxvirus to Enhance the Recognition of Tumor-Expressed Antigen
Preclinical Studies and Experimental Model Systems
We have used two preclinical models that we have developed and validated. The models grow progressively in their respective hosts, recapitulate expression of suppressive mechanism(s) found in clinical tumors, and express a characterized tumor or surrogate antigen for analysis. This latter point is critical. Relying solely on the generation of responses to whole tumor and tumor regression, although important, is complicated when one considers the overlapping regulatory mechanisms involved in the tumor–host environment. For this reason, we have developed model systems with well-characterized tumor antigens to which we have produced recombinant poxviral vaccines. These systems allow a more detailed in vitro and in vivo assessment of tumor antigen-specific responses.
MB49 (Transitional Cell Carcinoma of the Bladder)
MB49 was originally derived from cultures of male C57BL6 urothelium transformed in vitro with DMBA . MB49 grows subcutaneously as well as intravesically , where it responds to bacille Calmette–Guerin treatment in an analogous manner to human transitional cell carcinoma. In vivo growth of MB49 induces a high level of tumor-associated IL-10, resulting in the blockade of systemic T cell responses and inhibition of systemic DC function . Our earlier studies demonstrated that MB49 expresses the male-associated HY antigen complex encoded by a series of genes . Having this antigen has been critical in our ability to identify the previously mentioned regulatory mechanisms as well as generate an armamentarium of reagents (tetramers and peptides) that have allowed us to dissect the mechanism(s) of immune escape. We have also generated a series of recombinant vaccinia vaccines encoding the dominant MHC class I antigen encoded by the Uty gene (VV-Uty) and dominant MHC class II determinant encoded by the Dby gene (VV-Dby) , which have made our studies feasible and significantly add to our ability to define and manipulate mechanism.
NBT-1 (Transgenic-Derived Breast Tumor Model)
In addition to MB49, we have sought a nonchemically induced tumor model with characteristics that would make it more clinically relevant and at the same time more stringent vis-à-vis host recognition. To this end, we have derived and characterized a transplantable cell line that we have termed NBT-1 from a spontaneously arising mammary tumor from an MMTV-c-neu transgenic mouse made using a transforming neu construct pSV2neuNT . NBT-1 grows progressively in the nontransgenic female FVB parental strain of mice. NBT-1 is associated with VEGF and TGF-β 1–3 but not IL-10 mRNA (our unpublished results), which makes it an ideal target for our studies in that it expresses an overlapping but not identical panel of suppressive molecules to MB49. The tumor expresses the rat neu gene but grows progressively in both the parental nontolerant FVB mouse strain and the tolerant transgenic from which it was derived. We have also developed a recombinant vaccinia that encodes the full-length rat neu (VV-neu).
Preliminary Preclinical Analysis
As introduced previously, two potential areas of manipulation have been particularly important because they have been shown to be central to the development of all T cell responses. As indicated in Figure 17.1 , APCs are central to the induction of an effective T cell response to antigen. A critical step is the maturation and activation of antigen-presenting DCs. We were the first to demonstrate that IL-10 was overexpressed in primary biopsies of melanoma and bladder cancer . IL-10 is a potent negative regulator of DC maturation, is involved in the polarization of TH1 versus TH2 T cell responses , and has been associated with the activities of Treg cells in tumor systems . Using our MB49 tumor model, which recapitulate the tumor-associated IL-10 in patients, we demonstrated that IL-10 blocked the ability of tumor to prime for the expressed HY antigen , produced a TH2 polarizing immunization environment at the tumor site (immunization of B6 mice to bgal at the tumor site was skewed to TH2), and that these immune regulatory events were associated with or secondary to a significant impairment of DC function . (Splenic DCs from MB49-bearing mice manifested an IL-10-dependent depressed ability to stimulate allo-MLR and restimulate tumor antigen-specific T cells, and they were defective in antigen presentation when injected intratumorally into MB49 lesions ). All of these inhibited functions were restored in IL-10 knockout mice and/or when anti-IL-10 was administered prior to initial tumor challenge but not after the tumor had become established (our unpublished data) .
Also noted in Figure 17.1B , the highlighted factors associated with tumor growth have demonstrated abilities to modulate DC function directly or indirectly (IL-10, VEGF, and TGF-β) and/or be produced by tumor-associated DCs (IDO). Therefore, a critical focus of our studies has targeted DC maturation and function via modulating the previously mentioned factors and/or directly stimulating DC function in the tumor/DLN microenvironment and periphery. Our murine studies are supported by a clinical study demonstrating that DCs grown in the presence of IL-10 have reduced ability to stimulate MART-1-specific T cell responses unless transfected with antigen . These findings are consistent with those in a number of systems demonstrating the suppressive activity of IL-10 on DC function and the induction of DCs that stimulate Treg . This has led us to develop a strategy for enhancing the development of mature activated DCs with the ultimate goal of stimulating an optimal T cell response. Toward this end, we have used our vaccinia platform to transfer the GM-CSF gene to tumors in situ in both murine studies and translational clinical trials .
T Regulatory Cells Suppress Antitumor Immunity (The Balance between Effector and Regulatory Cells in the Tumor Microenvironment)
Ongoing studies from a number of laboratories have provided data supporting a major role for Treg cells in suppressing the development of antitumor immune responses in preclinical and clinical studies . Our recent studies have demonstrated an expansion of CD4 + CD25 + T cells in MB49 tumors and in the DLNs and that prophylactic depletion of Treg with in vivo anti-CD25 (PC61) resulted in rejection of MB49 challenge . Depletion after the tumor has become established has no direct antitumor effect but does convert mice from a systemically anergic phenotype (to tumor vaccine) to a vaccine-responsive phenotype (discussed later). These findings are consistent with studies from the Levitsky group that demonstrated that Tregs maintained a state of nonresponsiveness to tumor and importantly inhibited the ability to immunize with tumor antigen-specific vaccine likely due to a vaccine-induced expansion of Treg . In addition, studies from the Allison group have demonstrated a “balance” between effector and regulatory T cells in the tumor and DLN and an expansion of both populations following anti-CTLA4 treatment . Consistent with these findings, we have reported the presence of functional CTL in the DLN of MB49 , and concomitantly we have found antigen-specific Treg in the same compartment resulting in the mouse being functionally anergic to tumor antigen and systemically administered vaccine (our unpublished data).
Localized Immunotherapy of Tumors Optimizing Tumor Antigen Repertoire
Central to any immunotherapy/vaccine approach is the availability of relevant tumor-associated antigens. Although significant strides have been made in identifying tumor-specific antigens (TSAs), their numbers remain limited and focused on a few diseases. For this reason, we have chosen to use autologous tumor as an antigen source via local treatment strategies. Utilizing autologous tumor and our localized approach should provide multivalent antigens both shared and unique without requiring resection, the generation of autologous vaccines, or the identification of additional antigens. Our strategy of using the individual’s tumor as a source of antigen is further strengthened by the report that human colon and breast tumors harbor on average 90 individual mutations, all of which may represent immune targetable antigens . As noted previously, in addition to taking advantage of the range of tumor antigens expressed on tumor, focusing on the tumor microenvironment will ideally allow us to modulate both antigen presentation and T regulatory pathways critical to the development of a response.
In Vivo Murine Bladder Cancer Studies
To determine if recombinant vaccinia infects/transfects tumor, and perhaps normal mucosa, in vivo , the influenza hemagglutinin and nuclear protein encoding vaccinia recombinants (10 7 plaque-forming units (PFU)) were instilled via urethral catheters into the bladders of C57BL/6 mice bearing the MB49 tumor. After 8 h, the mice were euthanized, and their bladders were removed, sectioned, and stained for the two antigens immunohistochemically. When stained for the nuclear protein, we found substantial infection/transfection of the growing MB49 tumor . Although infected/transfected normal mucosa also stained for the encoded antigen, there was an apparent preferential staining in tumor. Whether this was due to a lack of protective glycosaminoglycan layer on the tumor or was peculiar to our early studies, it was clear that a high efficiency of infection/transfection of bladder tumor cells was obtained following intravesical instillation of the virus. There was seemingly no acute toxicity. Consistent with the preferential tumor localization of the recombinant are results from recent studies by the Bell group that showed preferential tumor localization following intravenous injection of recombinant vaccinia-GM-CSF (VV-GM-CSF) in murine models and in a phase I trial . Also supporting tumor localization was the group’s finding that the virus preferentially replicated in tumor cells expressing activated driver pathways .
Although we had shown that recombinant vaccinia infected bladder tumors in vitro and in vivo in naive animals, it was important to demonstrate infection/transfection in mice immune to the virus, which would be analogous to patients who had been vaccinated or following the first treatment. Intravesical administration of VV-NP (reporter gene construct) infects/transfects intravesically growing MB49 in the presence of systemic immunity. Mice were injected intraperitoneally with native vaccinia (shown to result in systemic anti-vaccinia immunity based on CTL generation). The mice were instilled with MB49 tumor intravesically, and 2 weeks later when tumor had been established they were instilled with VV-NP. Twelve hours later, the bladders were removed and stained immunohistochemically for the recombinant NP antigen that was positive. In addition, cytological changes including ballooning degeneration and intranuclear inclusion bodies were noted. Thus, systemic immunity to vaccinia that would be expected to be present in adult patients and following initial vaccinia treatments does not prevent intravesical tumor infection/transfection .
To demonstrate that recombinant vaccinia (VV) was able to recruit lymphocytes to the bladder wall and generate a systemic immune response, graded numbers of VV from 10 to 10 6 were instilled into bladders of C57BL/6 mice. Following 2 weeks of incubation, spleen cells were removed from the mice, restimulated in vitro with VAC for 7 days, and the resultant cells were tested for their ability to lyse the VAC-infected MB49 tumor target. As few as 10 PFU instilled intravesically resulted in significant VAC immunity, demonstrating its high degree of immunogenicity.
Clinical Translation of Preclinical Studies
The previously discussed preclinical studies have informed and led to the translation to a series of clinical trials focused on the feasibility and safety of injecting/instilling recombinant poxvirus intralesionally in patients with melanoma and bladder cancer.
Intralesional Vaccinia Vector in Patients with Melanoma
As a prelude to studying the effects of intralesional recombinant vaccinia in human melanoma, we obtained an Investigational New Drug application from the U.S. Food and Drug Administration to inject the Wyeth strain of vaccinia (the vaccine used in the United States for smallpox immunization and our nonrecombinant parent) intralesionally in patients with recurrent superficial melanoma . Following the demonstration of systemic immunity to vaccinia via an intradermal administration of vaccine to the patients, increasing doses of vaccinia were injected intratumorally. In a representative patient, 10 6 PFU of Wyeth vaccinia was injected into four sites in a 3-cm superficial melanoma lesion. The lesion was biopsied at 6 h and 4 days following administration. The biopsies were processed as frozen sections and stained using the monoclonal antibody TW2-3, which is specific for an early viral protein product of the EL3 gene present at sites of viral replication. A biopsy taken 6 h following intralesional vaccinia contained diffuse numbers of melanoma cells staining intracytoplasmically for the viral antigen . To determine if increasing immunity to vaccinia induced by multiple treatments would block productive infection/transfection, additional biopsies were similarly analyzed during the course of therapy in one patient who received 19 biweekly injections of as high as 10 7 PFU of virus (total cumulative dose of 13×10 7 PFU). Whereas the duration of expression was diminished with increasing immunity as measured by antiviral antibody titer, productive infection was seen throughout the treatment course . Note that minimal systemic side effects were seen in the trial. These findings demonstrate, as did our murine studies discussed previously, that vaccinia recombinants are able to infect/transfect tumor in vivo following intralesional injection even in the face of systemic immunity to the virus. It is our conclusion from these studies that systemic immunity to the virus acts to protect patients from toxicity while not preventing local gene expression. Our demonstration of sustained infection in virus-immune individuals strongly supports our approach in demonstrating that infection/transfection using cytokine gene-encoding vaccinia should result in cytokine production for a prolonged period.
Intralesional Vaccinia-GM-CSF Recombinant in Patients with Melanoma
Having been satisfied that the vaccinia vector met our requirements of safety and efficacy, we carried out a phase I trial of intralesional vaccinia-GM-CSF (produced in our laboratory and grown under good manufacturing practice conditions) in patients with therapy refractory recurrent melanoma . All patients were required to have accessible dermal and/or subcutaneous disease, with a number also having visceral disease. Following the demonstration of immune competence, which is important given the replicative nature of the vector, patients received twice-weekly intralesional injections of the recombinant with dose escalation within each patient. The results seen in the first seven patients are described in detail . At the highest doses, patients received 2×10 7 PFU per lesion, with the injection of multiple lesions resulting in as high as 8×10 7 per session. For comparison, vaccinia was used as a smallpox immunization at a scarification dose of 2.5×10 5 . Injection of VV-GM-CSF routinely led to the rejection of injected lesions and an associated influx of CD4 and CD8 T cells. In addition to the injected lesions, four of seven patients had regression of uninjected lesions following treatment . It was this latter regression of distant uninjected lesions that we have taken as evidence of the possible induction of tumor-specific immunity. Laboratory studies demonstrated that patients developed high levels of immunity to both vaccinia and the included β-galactosidase gene product . We also confirmed that in the face of maximal antibody titers, we continued to be successful at achieving recombinant gene expression as measured both as vaccinia-encoded GM-CSF (V-GM-CSF) (reverse-transcriptase polymerase chain reaction (RT-PCR) using primers designed to specifically identify viral encoded GM-CSF) and viral thymidine kinase gene (V-TK) mRNA expression .
Intravesical Vaccinia in Patients with Bladder Cancer
As a first step to our planned expansion of this strategy to the localized treatment of bladder cancer (our preclinical data demonstrated significant infection/transfection of the orthotopically growing murine bladder tumor MB49 following intravesical administration of recombinant vaccinia ), we completed a phase I study of intravesical vaccinia vector in patients with advanced transitional cell carcinoma . As with our phase I of vector alone in melanoma , we used the vaccinia in a dose-escalation study with each patient receiving three intravesical doses over a 2-week period. Given safety concerns, this study focused on patients with invasive transitional cell carcinoma scheduled for cystectomy, with the cystectomy scheduled for the day following the third dose. Patient characteristics have been reported , with doses employed and toxicity. As noted in our prior clinical trials, patients developed high titers of anti-vaccinia antibody, although maximal titers were measured after cystectomy given the shortened course of therapy. Also as noted previously, treatment was associated with a significant degree of inflammation and recruitment of activated T lymphocytes (CD3 + and CD45RO + ) .
Intravesical Instillation of Recombinant Fowlpox Virus Encoding GM-CSF or Tricom
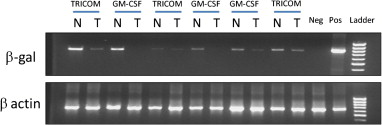
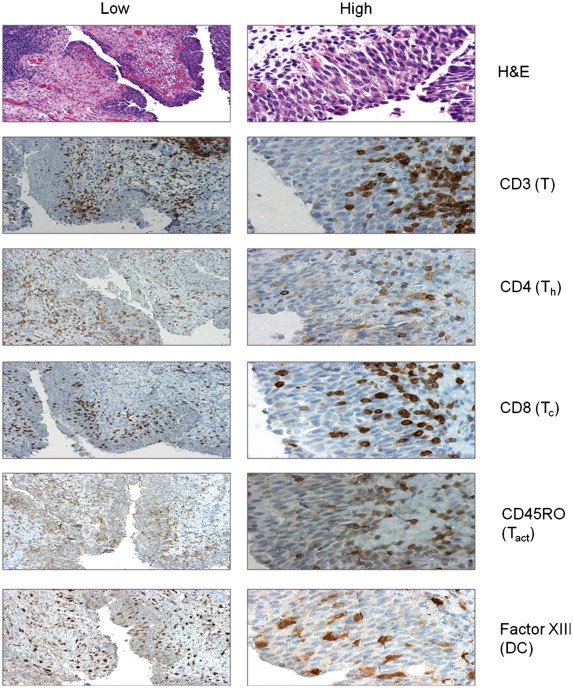
We recently expanded our GM-CSF studies to a study of intravesical fowlpox encoding either human GM-CSF or a three-gene construct made up of ICAM 1, LFA 3, and B7 developed at the National Cancer Institute (NCI) by the Schlom group and referred to as TRICOM . The study design was informed by the previously discussed intravesical vaccinia trial. The phase I study was supported by the NCI through the CTEP mechanism. We utilized nonreplicating fowlpox recombinants provided by the NCI. Patients scheduled for cystectomy for advanced bladder cancer received 4 weekly intravesical doses of recombinant fowlpox, with the last dose given 2 or 3 days prior to cystectomy. At the time of cystectomy, bladder tissue was collected and stained for a panel of immune cell antigens using immunohistochemistry. We also collected serum and PBMC to assess immune responses to the vector. In a similar manner as with our vaccinia-GM-CSF melanoma trial, the rF-GM-CSF and rF-TRICOM also encoded the lacZ gene, and infected cells/tissues could then be assessed for infection/transfection via RT-PCR for the lacZ gene product . Figure 17.2 demonstrates that bladder tissue from the treated patients expressed the β-galactosidase mRNA in both tumor and adjacent nontumor tissue, demonstrating successful infection/transfection. Immunohistochemical staining for a series of immune cell markers from a representative rF-GM-CSF patient is shown in Figure 17.3 . We found consistent infiltration of T cells of both CD4 and CD8 subsets in treated patients. The T cells were of the CD45 RO + activated phenotype. In addition, and consistent with our melanoma rV-GM-CSF study, we found significant infiltration of APC staining positively for factor XIIIa and representing DC populations ( Figure 17.3 ).
Related posts:
 Modified Oncolytic Herpesviruses for Gene Therapy of Cancer
Modified Oncolytic Herpesviruses for Gene Therapy of Cancer
 Genetically Engineered (T Cell Receptor) T Cells for Adoptive Therapy
Genetically Engineered (T Cell Receptor) T Cells for Adoptive Therapy
 Imaging of Oncolytic Virus Gene Expression
Ethics in Translational Gene Transfer Research
Imaging of Oncolytic Virus Gene Expression
Ethics in Translational Gene Transfer Research
 Viral Insertion Site Detection and Analysis in Cancer Gene Therapy
Viral Insertion Site Detection and Analysis in Cancer Gene Therapy
 Selectively Replicating Oncolytic Adenoviruses Combined with Chemotherapy, Radiotherapy, or Molecular Targeted Therapy for Treatment of Human Cancers
Selectively Replicating Oncolytic Adenoviruses Combined with Chemotherapy, Radiotherapy, or Molecular Targeted Therapy for Treatment of Human Cancers
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


