Fig. 8.1
Human VEGFA gene structure and exon composition of the isoforms generated by alternative splicing . (Top) The human VEGFA gene spans 16,272 bp of chromosome 6p21.1 and consists of eight exons and seven introns. In exon 1 red arrows show the start of the two promoter sites and black lines show the main translation start sites (AUG codons). In exon 8 the two alternative stop codons are indicated in red. (Bottom) Exon content of all known VEGF -A isoforms which are named according to the number of amino acids in the human protein. All isoforms contain exons 1–4 and one of two versions of exon 8 (8a or 8b) depending on which terminal splice site is used. Splice site 8a produces the VEGF-Axxx (or xxxa) pro-angiogenic isoforms and 8b produces the VEGF-Axxxb anti-angiogenic isoforms. Exons 6 and 7 encode heparin-binding domains so affect the diffusibility as an inverse relationship with their affinity for the extra cellular matrix
The most widely-studied physiological stimulus causing transcriptional up-regulation of the VEGFA gene is hypoxia . Hypoxic conditions initiate angiogenesis via VEGF -A throughout embryogenesis and during cancer progression. It is now well-established that the Hypoxia Inducible Factor (HIF) transcriptional activators are key mediators of the hypoxic response and the pathways leading to their activation are well-characterised. Both HIF-1 and HIF-2 bind to the VEGF-A hypoxia response element (HRE) (Forsythe et al. 1996; Blancher et al. 2000).
Since its discovery in 1989, the transcriptional regulation of VEGF -A has been extensively studied and a wide range of factors are known to be at play. However, more recent research has focused on the post-transcriptional regulation of VEGF-A, and this next stage of regulation has proven to provide yet more complexity.
3 VEGF -A Post-transcriptional Regulation
3.1 Alternative Splicing
The splicing process provides an opportunity for regulation of both the final protein structure and the level of protein expression. Different combinations of exons can be retained or removed to create a diverse array of mature mRNAs from the single pre-mRNA. In addition, alternative splicing within the non-coding regions of the mRNA controls the final number of regulatory elements such as translation enhancers or RNA stability domains that can significantly affect protein expression levels.
Through alternative splicing , nine isoforms of VEGF -A have been first found which are named according to the total number of amino acids in the mature proteins: 111, 121, 145, 148, 162, 165, 183, 189 and 206. In addition, some of these isoforms exist in VEGF-Axxxa or VEGF-Axxxb versions (Fig. 8.1). The VEGF-A gene contains eight exons. 1–4 are constitutive, being present in all isoforms, with exon 1 encoding an N-terminal signal of 26 hydrophobic amino acids that is typical of secreted proteins. Exons 5–8 are alternatively spliced to produce the different sized isoforms, with exons 6 and 7 containing basic residues that confer an affinity for heparin-binding. Heparin sulphate proteoglycans are present at the cell surface and within the extracellular matrix, thus the presence or absence of exons 6 and 7 affects the ability of that secreted VEGF-A isoform to diffuse away from the cell. In this way the presence or absence of exons 6 and/or 7 controls the spatial distribution and bioavailability of the VEGF-A isoforms (Vempati et al. 2011). As an example, VEGF-A121 lacks exons 6 and 7 thus does not bind heparin and is free to diffuse away after being released from the cell (Fig. 8.1). In contrast the VEGF-A165 and 189 isoforms are able to bind to heparin sulfate on the cell surface and in the extracellular matrix (Houck et al. 1992). These different heparin binding affinities result in the formation of a VEGF-A gradient which is essential for the process of angiogenesis, with soluble isoforms acting at distal sites to promote vascular recruitment and the extracellular membrane-associated isoforms acting locally to promote the expansion of capillary beds (Grunstein et al. 2000). The fact that an identical isoform can have distinct activities at different anatomical sites suggests that the microenvironment of individual tissues dictates VEGF-A function (Guo et al. 2001).
The C-terminus of VEGF -A is encoded by exon 8 and contains an alternative 3′ splice site that gives rise to the VEGF-Axxxa or VEGF-Axxxb versions of the isoform. Even if there is still controversy regarding the existence of the VEGF-Axxxb isoforms (Harris et al. 2012), in vitro and in vivo studies have demonstrated that the VEGF-Axxx (or -xxxa) isoforms are pro-angiogenic and are up-regulated in tumours whereas the VEGF-Axxxb isoforms are anti-angiogenic and are down-regulated in tumours (Bates et al. 2002; Woolard et al. 2004; Pritchard-Jones et al. 2007). To mediate their anti-angiogenic properties, the VEGF-Axxxb isoforms are thought to bind to the VEGF-A receptors but impair their downstream signaling (Woolard et al. 2004; Cebe Suarez et al. 2006), since when the VEGF-Axxxb and -xxx isoforms were co-expressed they acted in a dominant negative way and only partial receptor agonist activity was detected. This also suggests that the inhibitory function of the xxxb isoforms is mediated through competitive binding (Kawamura et al. 2008; Woolard et al. 2009). Further evidence for this has been found using recombinant human VEGF-A165b which possesses similar affinity towards the anti-Vascular Endothelial Growth Factor antibody (bevacizumab) as that of VEGF-A165, supporting the idea of inhibition through competitive binding. Interestingly, this also suggests that the balance between the expression of the anti-angiogenic and pro-angiogenic isoforms could regulate tumour growth and in the same way the anti-angiogenic xxxb isoforms could affect the sensitivity of tumours to bevacizumab through competitive binding (Varey et al. 2008). It was also demonstrated that the balance between the expression of the anti and pro-angiogenic isoforms could regulate follicle development (McFee et al. 2012), spermatogonial stem cell homeostasis in vivo (Caires et al. 2012) but also plays a critical role in the regulation of glomerular permeability (Oltean et al. 2012).
Recombinant human VEGF -A165b shows anti-angiogenic activity in eye hypoxia -driven angiogenesis (Konopatskaya et al. 2006). In addition, recombinant human VEGF-A165b possesses a similar affinity towards the anti-VEGF antibody (bevacizumab) as that of VEGF-A165, supporting the idea of inhibition through competitive binding . Interestingly, this also suggests that the balance between the expression of the anti-angiogenic and pro-angiogenic isoforms could regulate tumour growth and in the same way the anti-angiogenic xxxb isoforms could affect the sensitivity of tumours to bevacizumab through competitive binding (Varey et al. 2008).
The distal splice site used to generate the VEGF -Axxxb isoforms is 66 nucleotides further along the gene from the proximal splice site (Figs. 8.1 and 8.2) and results in a Ser-Leu-Thr-Arg-Lys-Asp C-terminal (Bates et al. 2002). This is very different from the Cys-Asp-Lys-Pro-Arg-Arg C-terminal generated by the proximal splice site in VEGF-Axxx isoforms and undoubtedly results in a distinct tertiary structure of the VEGF-Axxxb isoforms since they contain an acidic residue (Asp) in place of the Cys-160 present in the VEGF-Axxx isoforms, which forms a disulphide bond with the Cys-146 from exon 7. This was clearly proven for VEGF-A165 when the highly-charged C-terminal Pro-Arg-Arg tail of the VEGF-A165 isoform was replaced by the neutral Arg-Lys-Asp of the VEGF-A165b isoform and resulted in a profound alteration of the structure-function relationship of VEGF-A (Cui et al. 2004). VEGF-Axxxb isoforms have been identified for VEGF-A121, VEGF-A183, VEGF-A145, VEGF-165 (Perrin et al. 2005) and VEGF-A189 (Miller-Kasprzak and Jagodzinski 2008). VEGF-A165b was the first xxxb isoform identified and it is the most widely-studied (Bates et al. 2002). Its over-expression inhibits the growth of prostate carcinoma, Ewing’s sarcoma and renal cell carcinoma in xenografted mouse tumor models (Rennel et al. 2008) and inhibits tumor cell-mediated migration and the proliferation of endothelial cells (Rennel et al. 2008). Interestingly, mammary alveolar development during lactation is also inhibited by VEGF-A165b (Qiu et al. 2008) and endogenous VEGF-A165b contributes to survival of trophoblasts exposed to lower oxygen tensions through an autocrine pathway (Bills et al. 2014).
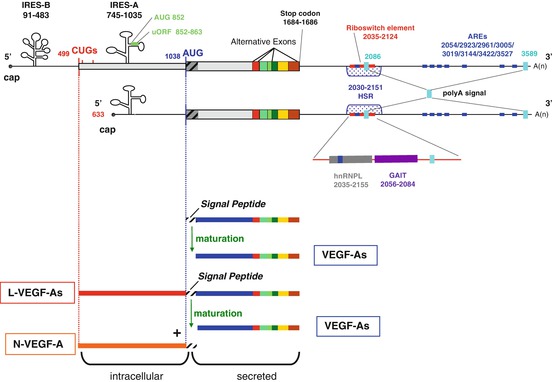
Fig. 8.2
Human VEGF -A mRNAs and their major regulatory elements. (Top) Schematic of VEGF-A mRNA transcribed from the two alternative promoters. Regulatory elements in the untranslated regions (UTR ) and the coding region are indicated; positions are numbered according to the mRNA sequence of the 5′ end of VEGF-A189. In the 5′UTR, two AUG start codons and three non-canonical CUG start codons are shown. IRES -B: internal ribosome entry site B, which controls transcription initiation from the CUG start codon. The IRES-A controls transcription initiation from the AUG start codons. uORF: upstream open reading frame, located in the IRES-A. In the 3′UTR, the two alternative polyadenylation sites are shown (polyA signal). AREs AU rich elements, HSR hypoxia stability region. The area around the riboswitch element has been expanded to show the binding sites for hnRNPL and the GAIT translation complex, which compete with each other for binding to this element in the regulation of oxidative stress . (Bottom) Initiation of translation at the CUG start codon produces the L-VEGF-A isoforms. These can be cleaved during the maturation process to yield an intracellular N-VEGF-A product and the mature secreted VEGF-A isoform that resembles the VEGF-A isoforms translated from the AUG start codon
Although many VEGF -A isoforms have now been identified, most VEGF-A-producing cells are thought to preferentially express VEGF-A121, VEGF-A165 and VEGF-A189. These are known as VEGF-A120, 164 and 188 in the mouse since there is one less amino acid in each mouse VEGF-A isoform. To clearly demonstrate the crucial role of alternative splicing in the regulation of VEGF-A activity, and to investigate the specific roles of the individual isoforms, transgenic mice have been generated that express only a single VEGF-A isoform. Mouse embryos expressing only VEGF-A120 (VEGFA 120/120 ) show impaired post-natal cardiac angiogenesis, resulting in severe myocardial ischemia, early post-natal death and impaired lung vascular development (Mattot et al. 2002). Half of the embryos die in the perinatal period due to congenital birth defects, with the other half perishing within 2 weeks after birth, in part due to myocardial ischemia (Carmeliet et al. 1999). Similar studies with mice expressing only VEGF-A188 (VEGFA 188/188 ) also show significant effects, with half of all embryos dying between embryonic stage E9.5 and E13.5 (Stalmans et al. 2002). Interestingly, mice expressing only VEGF-A164 (VEGFA 164/164 ) are healthy (Stalmans et al. 2002).
Together these transgenic mouse studies suggest that different alternatively spliced isoforms are required at different stages in normal development. Other findings support this since it has been shown that different isoforms are expressed in distinct spatio-temporal patterns both during embryonic development and in adult tissues (Ng et al. 2001). However, it is also known that there is some functional redundancy between isoforms, for example during the initial formation of arch arteries (Stalmans et al. 2003).
The importance of alternative splicing of VEGF -A pre-mRNA in terms of VEGF-A regulation is now well-established. Despite this, very little is known about the mechanisms regulating alternative splicing and thus how the cell controls the levels of the different VEGF-A isoforms. Exonic splicing enhancer/silencer sequences are sequences of DNA that promote/reduce the recognition and use of splice sites. An exonic silencer sequence (ESS) has been proposed to be present in exon 6 of VEGF-A, but the regulatory proteins interacting with this sequence remain unknown (Wang et al. 2009). In the alternative splicing process, Serine/Arginine rich proteins (SR proteins) also play a key role in identifying splice sites. Under hypoxic conditions the expression and phosphorylation of the SR proteins ASF/SF2, SRp20 and SRp40 were shown to correlate with increased VEGF-A expression and a shift towards expression of the VEGF-A121 isoform in the endometrial cancer cell line RL95 (Elias and Dias 2008). Increased expression of VEGF-A121 over the longer isoforms is known to be mediated by two splicing factors of the U2AF65 protein family, namely CAPERalpha and CAPERbeta (Dowhan et al. 2005). SR proteins have also been implicated in the alternative splicing of exon 8, with the ASF/SF2 and SRp40 proteins favoring the use of the pro-angiogenic proximal splice-site, and the SRp55 protein promoting the use of the anti-angiogenic distal splice-site (Nowak et al. 2008). However, other factors are also known to play a role in the pro-/anti-angiogenic decision: the E2F1 transcription factor has been shown to favor the expression of anti-angiogenic VEGF-Axxxb isoforms through a mechanism involving the expression of the splicing factor SC35 (Merdzhanova et al. 2010), and now a link between the Wilms’ tumour suppressor gene (WT1) and the regulation of VEGF-A pro-/anti-angiogenic alternative splicing has been established (Amin et al. 2011). WT1 binds to the promoter region of SRPK1 (Serine/Arginine-rich protein-specific kinase 1), a kinase that regulates the activity of SR proteins. This decreases the expression of the kinase, resulting in decreased AFS/SF2 phosphorylation and nuclear localization, and an increase in expression of anti-angiogenic VEGF-A165b. Conversely, mutation of the WT1 tumor suppressor gene results in ASF/SF2 hyperphosphorylation and expression of pro-angiogenic VEGF-A isoforms (Amin et al. 2011). These data suggest that tumors lacking functional WT1 or containing enhanced SRPK1 expression may control angiogenesis through the regulation of alternative splicing of VEGF-A.
There is no doubt that our understanding of the mechanisms regulating alternative splicing is still in its infancy. A recent study proved that the alternative splicing field still has many more doors to open when it described a novel VEGF -A isoform in the lung tissue of a legally aborted female fetus. This alternatively spliced isoform possessed a 20 bases insertion in the third intron, inducing a frame shift mutation that introduced a stop codon in the middle of the fourth exon (Zhou et al. 2012). The mechanism involved in this splicing regulation and whether this alteration impact VEGF-A protein structure and/or function is still unknown but this discovery highlights the discrepancies in our understanding of this important process.
Reinforcing this biological control, a VEGF-A stop-codon readthrough mechanism has been recently discovered that leads to a 22 amino-acids extension generating new antiangiogenic isoforms (Eswarappa et al. 2014).
3.2 mRNA Stability
In cells different mRNAs have very different stabilities. The more stable an mRNA is the longer its lifetime and hence the more protein is produced from it. Short mRNA half-lives allow cells to alter protein synthesis rapidly when necessary (for example in response to environmental changes such as nutrient levels, cytokines, hormones or stress ) and in this way the control of mRNA stability is an important regulator of protein expression (Mitchell and Tollervey 2000; Shim and Karin 2002).
VEGF -A mRNA is highly unstable under normal oxygen and nutrient conditions with a half-life of 15–40 min in vitro (Dibbens et al. 1999; Ikeda et al. 1995; Levy et al. 1996; Shima et al. 1995). Hypoxic conditions induce VEGF-A expression by increasing mRNA stability through a regulatory process that is independent of HIF1A-induced transcription (Ryan et al. 2000). Like most mRNAs with a short half-life, VEGF-A mRNA contains many AU-rich elements (AREs) which are well-known cis-sequences generally composed of different nonameric (AUUUA) or pentameric (AUUUAUUUA) consensus or U-rich sequences. They increase the rate of degradation of the mRNA and so by blocking or promoting access to these sites the cell can control the stability of the mRNA (Barreau et al. 2005; Gingerich et al. 2004). The VEGF-A AREs are clustered in the 3′UTR section of VEGF-A mRNA (Fig. 8.2). In humans, the stability of VEGF-A mRNA under hypoxic conditions is mediated through the 3′UTR (Claffey et al. 1998; Goldberg-Cohen et al. 2002; Levy et al. 1998), therefore research on the control of VEGF-A expression in response to hypoxia has focused on ARE -binding proteins. Interestingly, it has been shown that degradation of mouse VEGF-A mRNA under normoxic conditions requires the independent action of destabilizing elements from across the mRNA (in the 3′UTR, the coding region and the 5′UTR). In contrast to human VEGF-A mRNA, under hypoxic conditions mouse VEGF-A mRNA requires all three sections and these coordinate to stabilize the mRNA (Dibbens et al. 1999). In humans a number of RNA-binding proteins have been shown to interact with the 3′-UTR elements of VEGF-A mRNA and increase its stability including: the CSD/PTB complex (Cold Shock Domain/Polypyrimidine Tract Binding Protein) (Coles et al. 2004); MDM2, a protein translocated from the nucleus to the cytoplasm under hypoxic conditions (Zhou et al. 2011); hnRNPL, which associates with hnRNP complexes to promote mRNA formation, processing and packaging (Shih and Claffey 1999); the double strand RNA binding protein DRBP76/NF90, which under hypoxic conditions facilitates VEGF-A expression by promoting mRNA loading onto polysomes and translation (Vumbaca et al. 2008); HSP70, which was found to bind and stabilize VEGF-A mRNA through a mechanism independent of its chaperone function (Kishor et al. 2012); and HuR (Levy 1998). HuR is a member of the ELAV protein family, which also includes the Hel-N1, HuC and HuD proteins (Fan and Steitz 1998; King 2000; Levine et al. 1993; Peng et al. 1998). ELAV proteins bind to AREs and are thought to regulate the stability of mRNA from splicing through to translation by generating multimeric “ribonucleosomes” due to their nucleation and cooperative binding properties. For example, HuR bound to the 3′UTR has been shown to interact directly with PAIP2 (Poly(A)-Binding Interacting Protein 2) that is bound to a distinct region of the mRNA, and it has been suggested that interaction between these proteins can influence the overall RNA structure and render it inaccessible to endonucleases (Onesto et al. 2004, 2006). Recently it was also proposed that ELAV/Hu proteins can displace miRNAs (which would otherwise silence the mRNA; see Sect. 3.4.3) and thus suppress their destabilizing properties (Simone and Keene 2013). This seems to be the case for mouse VEGF-A mRNA since the HuR and miR-200b binding sites overlap and it was shown that the miR-200b-induced suppression of VEGF-A expression was competitively antagonized by HuR at the 3′UTR (Chang et al. 2013). Finally, HuR has also been implicated in effective VEGF-A mRNA export from the nucleus and loading onto active polysomes under hypoxia. Along with hnRNPL and hnRNPA1, the extra-nuclear shuttling of these mRNA-binding proteins has also been shown to regulate VEGF-A mRNA stability (Vumbaca et al. 2008).
The 3′UTR ARE of VEGF -A mRNA can also be the target of proteins that destabilize mRNA in various mammalian cell types, for example AUF1 and tristetraprolin (TTP ) (Bernstein and Ross 1989; DeMaria and Brewer 1996; Gorgoni and Gray 2004; Stoecklin et al. 2003; Zhang et al. 2002). AUF1 promotes the assembly of other factors necessary for recruiting the mRNA degradation machinery, such as translation initiation factor eIF4G, heat-shock cognate protein hsc70, lactate dehydrogenase and the poly(A)-binding protein. Interestingly, although the poly(A)-binding protein is a stabilizing factor for polyadenylated mRNAs, it has destabilizing effects on VEGF-A mRNA (Gorgoni and Gray 2004; Ma et al. 2006). It was recently shown that AUF1-mediated regulation of VEGF-A expression in mouse macrophage-like RAW-264.7 cells is mediated through its C-terminus domain that contains a region with three arginine-glycine-glycine (RGG) motifs (Fellows et al. 2012). AUF1 itself can be regulated by many signaling pathways and therefore is subjected to numerous post-translational modifications such as phosphorylation, glycosylation, methylation and ubiquitination (for a review, see Gratacos and Brewer 2010). TIS11/Tristetraprolin (TTP) is another ARE-binding protein that regulates the destabilization of VEGF-A mRNA through a signaling pathway involving casein kinase 2 and p38 MAPK (Lee et al. 2011). These examples show the breadth of cellular signaling pathways involved in the regulation of VEGF-A mRNA stability and the depth of their control by the cell.
3.3 Alternative Polyadenylation
Polyadenylation is the addition of a poly(A) tail to the 3′UTR of an mRNA and is important for mRNA stability , nuclear export and translation. Before the poly(A) tail is added, the 3′end of the mRNA is cleaved at specific sites, thus if an mRNA contains more than one polyadenylation site this can produce alternative transcripts, as for alternative splicing .
Although the 3′UTR is known to play a pivotal role in the post-transcriptional control of VEGF -A expression (Bartel 2009), our knowledge of the use and regulation of poly(A) signals is very limited. VEGF-A cDNA sequencing has revealed the presence of two major polyadenylation sites: a consensus AAUAAA site and a non-canonical AUUAAA site, located 399 and 1902 nucleotides after the stop codon respectively (Fig. 8.2) (Claffey et al. 1998; Dibbens et al. 2001). The resulting mRNAs from both sites have been characterized (Leung et al. 1989; Keck et al. 1989), but only one study has investigated the use of VEGF-A alternative polyadenylation (Dibbens et al. 2001). Using a mouse model it reported that the majority of VEGF-A transcripts are processed at the distal polyadenylation site (resulting in mRNAs containing the longer form of the 3′UTR), and that the same site is used under both hypoxic and normoxic growth conditions (Dibbens et al. 2001). Given the large section of the regulatory 3′UTR that is missing in transcripts using the proximal polyadenylation site, it is possible that the resulting VEGF-A isoforms play distinct roles. Therefore, much more work is needed to ascertain the importance of this method of VEGF-A regulation.
3.4 Translational Regulation
3.4.1 Internal Ribosome Entry Site (IRES ) Elements and Alternative Initiation at Non-AUG Codons
In eukaryotic cells translation usually depends on the presence of an m7G cap at the 5′ terminus of the mRNA. The cap is required for assembly of the initiation complex, and allows translation to begin at the start codon further downstream. Traditionally the start codon is an AUG coding for methionine; however, alternative start codons are now known to exist, such as CUG which codes for leucine and whose initiation pathways are thought to be independent of those of the AUG codon (Starck et al. 2012). In addition, translation of a number of cellular mRNAs can be initiated from the middle of an mRNA sequence through a cap-independent mechanism. These mRNAs contain an internal ribosome entry site (IRES ) within their 5′ UTR . IRESs are structural elements that are specifically required to maintain and/or activate the expression of specific proteins during cell stress situations when cap-dependent translation is compromised (Spriggs et al. 2008). They are thought to have an extensive predicted secondary structure and have been reported mostly in mRNAs containing long 5′UTRs with a high GC content. To date more than 70 viruses and more than hundred eukaryotic mRNAs are known to contain at least one IRES, including VEGF -A (Mokrejs et al. 2010).
VEGF -A mRNAs have a 1038-nucleotide-long GC-rich 5′UTR upstream of the classical AUG start codon which contains three in-frame alternative CUG start codons and two IRESs (Fig. 8.2) (Akiri et al. 1998; Huez et al. 1998; Miller et al. 1998). The first IRES to be identified, IRES-A, lies within the 300 nucleotides upstream from the AUG start codon and controls the initiation of translation at this point (Fig. 8.2). IRES-B is located closer to the 5′cap, 16 nucleotides upstream of the first CUG codon and controls initiation from the two alternative CUG start codons. This IRES-mediated control of AUG versus CUG has been demonstrated both in vitro (Akiri et al. 1998; Huez et al. 1998) and in vivo (Bornes et al. 2007). In vivo, VEGF-A IRESs are thought to be particularly important for responding to local environment stresses where cap-dependent translation is inhibited, such as hypoxia , and indeed it has been shown that IRES have a low activity in embryos and adult tissues but allow efficient translation at early time points in ischemic muscle (Bornes et al. 2007). Recent studies have now focused on identifying the regulatory factors involved in IRES-mediated translation. One study used a high-throughput screening approach that combined siRNA treatment with transfection of a VEGF-A IRES reporter mRNA, to identify and validate MAPK3 kinase as a novel positive regulator (Casanova et al. 2012). The DEAD-box RNA helicase 6 (DDX6) has also been identified as an IRES trans-acting factor. Using MCF-7 cell line extracts it was shown that under normoxic conditions recombinant DDX6 inhibits VEGF-A IRES-mediated translation whereas hypoxic conditions caused a decrease in DDX6 levels that led to the induction of VEGF-A expression (de Vries et al. 2013). VEGF-A IRES can also be controlled from within the mRNA and in this way they have been shown to play a role in dictating the use of the alternative start codons, AUG and CUG. For example, mRNA encoding the alternatively spliced VEGF-A165 and -189 sequences can be efficiently expressed through initiation events using both start codons. The choice of initiation site is determined through modulation of IRES-A activity by the alternatively spliced coding sequences (Bornes et al. 2004). It is thought that different splice variants contain different elements that, through long-range interactions within the VEGF-A mRNA, could regulate IRES-A activity to promote/inhibit the use of the AUG site, however the exact molecular mechanism of this control is still unknown.
Initiation of translation from the CUG codon is equally efficient regardless of which splice variant is expressed (Bornes et al. 2004). The first CUG start codon is located 539 nucleotides upstream of the coding sequence (Huez et al. 2001; Meiron et al. 2001; Tee and Jaffe 2001; Touriol et al. 2003). Initiation of translation at this CUG codon leads to the production of longer VEGF -A isoforms (L-VEGF-A) containing an additional 180 amino acids (Fig. 8.2). During maturation, L-VEGF-A can be cleaved at the peptide signal sequence to generate both a shorter secreted VEGF-A isoform and an intracellular portion termed N-VEGF-A. Interestingly, the VEGF-A121 isoform is known to be exclusively expressed through initiation at the CUG codon and thus is a maturation product of L-VEGF-A121. Following L-VEGF-A cleavage, the N-VEGF-A fragment is released and translocates to the nucleus (hence the “N” prefix). It is a 23-kDa NH2-specific peptide containing 206 amino acids which are highly conserved among mammals, suggesting that N-VEGF-A has an important function, the details of which are yet to be identified (Fig. 8.2) (Huez et al. 2001; Tee and Jaffe 2001; Rosenbaum-Dekel et al. 2005). The role and purpose of L-VEGF-A is also not well-understood although it has been suggested that it may serve as a reservoir for the generation of shorter isoforms since an increase in the longer intracellular L-VEGF-A165 and L-VEGF-A189 protein isoforms has been linked to a reduction in shorter secreted isoforms (Chiarini et al. 2006). Despite our lack of understanding of the role of L-VEGF-A, its importance is demonstrated by a single nucleotide polymorphism in the human VEGF-A gene (-634 C-G) that causes IRES -B dysfunction and leads to a 17 % reduction in initiation at the CUG codon and thus a decrease in L-VEGF-A expression (Lambrechts et al. 2003). This polymorphism has been associated with a number of serious conditions, such as an increased risk of motor neuron degeneration in amyotrophic lateral sclerosis (ALS) (Lambrechts et al. 2003), the development of diabetic macular edema that correlates with macular retinal thickness in type 2 diabetes (Awata et al. 2005), an increased aggressiveness of breast cancer (Jin et al. 2005), and an increased risk of gastric (Guan et al. 2009) and prostate cancer (Sfar et al. 2006). These studies not only provide evidence of the crucial role of IRES function and the significance of the L-VEGF-A isoforms but also illustrate how these distinct regulatory elements are interdependent such that a single mutation in one element has such a serious downstream effect on many others.
3.4.2 RNA G-Quadruplex Structure
G-quadruplexes are guanine-rich regions of the mRNA that can fold up to form secondary structures organized in stacks of planar layers of guanine tetrad (or quartet) units. It is emerging that these elements have regulatory functions in different steps of RNA metabolism, including mRNA translation (Millevoi et al. 2012). G-quadruplexes are found in coding and non-coding mRNA. The majority of those that are located in 5′UTRs play an inhibitory role in cap-dependent translation (for example in Zic-1, ESR1, NRAS or MT3-MMP mRNA), probably through the recruitment of stabilizing proteins to prevent ribosome scanning (Bugaut and Balasubramanian 2012).
VEGF -A mRNA contains a 17 nucleotide-long element within the IRES -A site (nucleotides 774-790) that can fold into a two-G-quartet quadruplex structure. It is thought that this is important for IRES-A function since IRES-A activity was blocked by mutations that disrupted the intramolecular G-quadruplex structure (Morris et al. 2010). This finding is quite surprising since most naturally-occurring G-quadruplexes with the ability to regulate translation are at least three-G-quartet quadruplex structures while artificially engineered two-G-quartet quadruplexes like that found in VEGF-A have shown only a moderate stability (Bugaut and Balasubramanian 2012). Thus, it is important to establish whether the VEGF-A G-quadruplex contributes to IRES-A activation under stress conditions such as hypoxia , endoplasmic reticulum stress or ischemia, since this is when the IRESs are active and required for the initiation of translation. If this structure affects IRES-A activation under these conditions then G-quadruplex formation will be confirmed as yet another mechanism of VEGF-A regulation.
3.4.3 Upstream Open Reading Frame (uORF)
Upstream open reading frames (uORFs) are generally short sequences located within the 5′UTR of an mRNA. They can regulate protein translation under normal and stress conditions, usually by inhibiting expression of the main transcript (Calvo et al. 2009; Spriggs et al. 2010). Generally, uORFs act as a constitutive barrier to the scanning ribosome and thereby reduce ribosome access to the main AUG codon. uORFs are found in the 5′UTR of proto-oncogenes, cytokines and many other eukaryote mRNAs, including human 5-HT3A (5-hydroxytryptamine receptor 3A), TPO, BACE1 [β-site APP (Amyloid Precursor Protein) cleavage enzyme 1] and the huntingtin protein (the protein associated with Huntington’s disease) (Chatterjee and Pal 2009).
The VEGF -A 5′UTR has a unique, short uORF that is highly-conserved between species and begins at AUG 852, 186 nucleotides upstream of the main AUG start codon (Fig. 8.2). It is located within the IRES -A sequence and is translated through a cap-independent mechanism. It has been suggested that the VEGF-A uORF acts as a cis-regulatory element involved in the shut-off of the CUG codon in the control of VEGF-A121 expression since mutation of AUG 852 (Fig. 8.2) increased translation of the VEGF-A 121 isoform but had no effect on the expression of the VEGF-A165 or -189 isoforms (Bastide et al. 2008). VEGF-A121 translation occurs exclusively downstream of the CUG start codon, thus an active uORF could inhibit its translation and in that way contribute towards controlling the expression of the different VEGF-A isoforms. As a potentially constitutive inhibitor, the VEGF-A uORF may be regulated by specific sequences, trans-acting factors or conditions that have not yet been identified due to the difficulties in predicting the effect of uORF alterations. Despite this, the VEGF-A uORF clearly affects its expression and is another factor contributing to its fine-tuning.
3.4.4 miRNA-Mediated Regulation of VEGF -A
MicroRNAs (miRNAs ) consist of a small (20–25 nucleotides) sequence of non-coding RNA that control the translation of genes through RNA silencing. They are generated from local hairpin structures by two RNA endonucleases, Drosha and Dicer (Kim et al. 2009). The importance of miRNA in VEGF -A regulation was first suggested when researchers mutated the dicer gene in mice. This led to retarded development, defective angiogenesis and the death of embryos between days 12.5 and 14.5 of gestation. This phenotype correlated with over-expression of VEGF-A, therefore it was concluded that the defect in angiogenesis was most likely due to a deficiency in VEGF-A mRNA processing (Yang et al. 2005). Since then many miRNAs have been identified that target VEGF-A mRNA, all of which bind to the 3′UTR region. Figure 8.3 summarizes the different miRNA binding sites found. Close to the end of the main ORF is a binding sequence for miR-125a, which has been shown to inhibit the proliferation and metastasis of hepatocellular carcinoma by targeting VEGF-A (Bi et al. 2012). This is followed by a sequence for miR-205, whose expression in glioma cell lines increases VEGF-A expression (Yue et al. 2012). Further along is a binding site for a number of miRNAs, including miR-20a and miR-20b. These were some of the first VEGF-A-binding miRNAs to be identified and, like miR-15b and miR-16, they were found to co-regulate other angiogenic factors in addition to VEGF-A regulation (Hua et al. 2006; Lei et al. 2009). miR-93b also binds to this site; it was shown to be down-regulated under hyperglycemic conditions which correlated with increased VEGF-A expression (Long et al. 2010). The miR-16 responsive element is further along, located 270 nucleotides downstream of the translation stop codon (Karaa et al. 2009), and affects angiogenesis in multiple myeloma (Sun et al. 2013). Interestingly, miR-195, which was demonstrated to suppress angiogenesis and metastasis of hepatocellular carcinoma (Wang et al. 2013), overlaps the miR-16 binding site, suggesting a mutually exclusive interaction of these 2 miRNAs. Close to the first poly(A) site lies a CA Rich Element (CARE) which is targeted by at least four miRNAs, miR-297, miR-299, miR-567, and miR-609. Their binding can induce a robust inhibition of protein synthesis under normoxic conditions (Jafarifar et al. 2011). miR-145 and miR-199a-5P then binds to an overlapping sequence further downstream. Through its post-transcriptional regulation of VEGF-A in endometrial mesenchymal stem cells miR-199a5P contributes to the pathogenesis of endometriosis (Hsu et al. 2014) while miR-145 has been shown to inhibit tumor angiogenesis, invasion and growth (Zou et al. 2012; Fan et al. 2012). Following this are sites for miR-126 and miR-200b binding; their respective down-regulation in lung cancer cells and in the diabetic retina cause increased VEGF-A expression (Liu et al. 2009; McArthur et al. 2011). By targeting a sequence located just downstream of the miR-200b binding site, miR-377 regulates mesenchymal stem cell-induced angiogenesis in ischemic hearts (Wen et al. 2014). Finally, the miR-361-5p, miR-29a, miR-29b and miR-190-binding sites are close to the second poly(A) site. MicroRNA miR-361-5p level is inversely correlated with VEGF-A expression in human cutaneous squamous cell carcinoma (Kanitz et al. 2012), and miR-29a, miR-29b and miR-190 were demonstrated to repress metastasis and angiogenesis (Chen et al. 2014; Melo and Kalluri 2013; Hao et al. 2014).
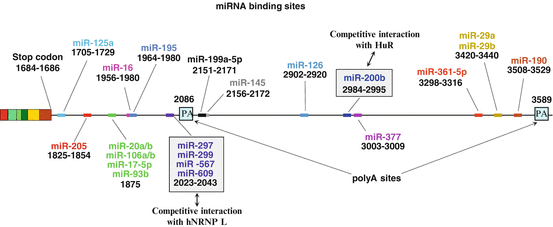
Fig. 8.3
miRNA-targeting sites in the human VEGF -A mRNA 3′UTR . The 3′ untranslated region of human VEGF-A mRNA is shown with the two alternative polyadenylation (PA) sites. The position of each miRNA target site is illustrated and numbered according to the 5′end of the VEGF-A 189 mRNA sequence
At least two of the miRNA binding sites are known to control VEGF -A expression through competitive binding with regulatory proteins. The RNA binding protein HuR , which plays a role in mRNA stability (see Sect. 3.2), can also bind to the same site as miR-200b. Their competitive binding has been shown to control VEGF-A expression in bone marrow-derived macrophages (Chang et al. 2013) in a mechanism that is evolutionarily conserved, at least between mouse and zebrafish. At a separate location, the hnRNPL protein can bind to the CARE sequence of the 3′UTR when it is delocalized from the nucleus to the cytoplasm under hypoxic conditions. This presumably renders this element inaccessible to the RNA-Induced Silencing Complex with its incorporated miRNAs (the miRISC), which target the same element (Jafarifar et al. 2011). This mechanism also demonstrates the key role of hnRNPL during hypoxia since it is also able to promote VEGF-A mRNA stability (see Sect. 3.2) and plays a role in the VEGF-A mRNA riboswitch (see below).
Related posts:
 Translational and Post-translational Control of Leptin Production by Fat Cells
Translational and Post-translational Control of Leptin Production by Fat Cells
 MicroRNA Regulation of Endocrine Functions in the Ovary
MicroRNA Regulation of Endocrine Functions in the Ovary
 Post-transcriptional and Post-translational Regulation of Steroidogenesis
Post-transcriptional and Post-translational Regulation of Steroidogenesis
 Post-transcriptional Regulation of Insulin and Insulin Like Growth Factors
Post-transcriptional Regulation of Insulin and Insulin Like Growth Factors
 Mechanisms of Post-transcriptional Gene Regulation
Mechanisms of Post-transcriptional Gene Regulation
 Post-transcriptional Regulation of Parathyroid Hormone Gene Expression in Health and Disease
Post-transcriptional Regulation of Parathyroid Hormone Gene Expression in Health and Disease
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


