Fig. 9.1
Overview of the PGE2 synthesis pathway. Arachidonic acid is liberated from phospholipids by phospholipase A2 and phospholipase C activity. Free arachidonic acid can then be converted into the intermediate PGH2 by either isoform of the cyclooxygenase enzymes, COX-1 and COX-2. PGE synthases cPGES, mPGES-1, and mPGES-2, convert PGH2 into PGE2 that signals through the G protein-coupled receptors EP1, EP2, EP3, and EP4 in both an autocrine and paracrine manner
1.3.1 PGE2
PGE2 is the most abundant prostaglandin associated with pathologies involving inflammation, pain sensitivity and fever, and neoplasia (Park et al. 2006; Kawahara et al. 2014; Wang and Dubois 2010). PGE2 also promotes gastric mucosa protection, renal hemodynamics, and stimulates ovulation and myometrium contraction during parturition (Takeuchi 2010; Stouffer et al. 2007; Yount and Lassiter 2013). Three PGE2 synthases (PGES), cytosolic cPGES and two membrane-associated PGES, mPGES-1 and the microsomal mPGES-2, produce PGE2 by using endoperoxide PGH2 as a substrate (Regan 2003). mPGES-2 and cPGES are constitutively expressed, whereas mPGES-1 is induced by pro-inflammatory cytokines (Kudo and Murakami 2005; Ricciotti and FitzGerald 2011; de Oliveira et al. 2008). cPGES interacts with COX-1 and preferentially converts COX-1-dependent PGH2, whereas mPGES-1 is the major enzyme involved in PGE2 production from COX-2 derived PGH2 (Regan 2003; Kudo and Murakami 2005). mPGES-2 is produced as a golgi membrane-associated protein and is associated with both COX-1 and COX-2 (Kudo and Murakami 2005).
PGE2 signaling is mediated via the four membrane receptors EP1, EP2, EP3, and EP4 encoded by PTGER1, PTGER2, PTGER3 and PTGER4 genes, respectively (Funk 2001; Hull et al. 2004; Breyer et al. 2001; Sugimoto and Narumiya 2007). EP receptors are G protein-coupled receptors primarily localized to the cell surface, although evidence exists indicating nuclear membrane localization (Bhattacharya et al. 1998; Breyer and Breyer 2000; Funk 2001). EP1 is a Gαq-coupled receptor coupled phospholipase C/inositol triphosphate signaling and free Ca2+ mobilization (Breyer et al. 1996). EP2 and EP4 are Gαs-coupled receptors that stimulate adenylate cyclase to produce cAMP, which in turn activates kinases such as protein kinase A (Hull et al. 2004; Regan 2003). EP3 receptor consists of multiple splice variants and has been shown to couple to both Gαi and Gαs proteins, leading to reduced and increased cAMP levels, respectively (Namba et al. 1993; Kotani et al. 1997; Hatae et al. 2002).
The action of PGE2 in a tissue-specific physiology predominantly depends on the cell-specific EP receptor expression and levels of the four receptor subtypes (Breyer et al. 2001). EP3 and EP4 receptors are the most widely expressed subtypes, observed in all tissues examined (Sugimoto and Narumiya 2007). Based on observations using pharmacological antagonists and genetic knockout studies of EP receptors, it is of considerable interest to further explore their post-transcriptional regulation in order to control their expression levels (Kawamori et al. 2001; Yang et al. 2006; Keith et al. 2006).
1.3.2 PGI2
PGI2 (prostacyclin) is produced by PGI2 synthase (PGIS) primarily in vascular endothelial cells and smooth muscle cells for maintenance of cardiovascular homeostasis (Dorris and Peebles 2012; Ricciotti and FitzGerald 2011). The biological effects of PGI2 are mediated through the Gαs-coupled IP receptor, whose expression has been found in platelets, heart, aorta, lung, kidney, and liver (Dorris and Peebles 2012). Similar to EP2 and EP4 receptors, the IP receptor promotes activation of adenylate cyclase and PKA (Dorris and Peebles 2012). PGI2 is also involved in physiological functions such as gastric mucosa protection, smooth muscle relaxation, and vasodilatation (Williams et al. 1994; Harada et al. 1999; Tanaka et al. 2004). Moreover, PGI2 is a potent inhibitor of platelet aggregation, leukocyte adhesion, and vascular smooth muscle cells proliferation (Dorris and Peebles 2012). In cancer cells, PGI2 stimulates angiogenesis, through IP receptor expressed at the surface of tumor endothelial cells (Osawa et al. 2012). It has also been shown that PGI2 can signal through the nuclear receptor PPARδ (peroxisome proliferator-activated nuclear receptor) and PGI2-induced PPARδ activation can regulate arterial thrombus formation, blastocyst development, and limits inflammation (Kang et al. 2011; Barbieri et al. 2012; Chen et al. 2009; Dorris and Peebles 2012).
1.3.3 PGD2
PGD2 is mainly synthesized in the CNS, mast cells, dendritic cells, macrophages, and Th2 lymphocytes (Ricciotti and FitzGerald 2011). PGD2 is implicated in platelet aggregation, smooth muscle contraction, broncho-constriction, immune cells chemotaxis, and regulation of sleep and wake cycles (Urade and Hayaishi 2011; Kanaoka and Urade 2003). PGD2 is synthesized by two enzymes PTGDS and HPGDS. PTGDS is mainly expressed in brain tissues, male genital organs and in the heart (Urade and Hayaishi 2000), where HPGDS is hematopoietic in origin (Kanaoka and Urade 2003). PGD2 plays a pro-inflammatory role through binding to the DP receptors DP1 and DP2 encoded by PTGDR1 and PTGDR2 genes, respectively. DP1 is a Gαs-coupled receptor ubiquitously expressed and the activation of this receptor leads the activation of adenylate cyclase and PKA (Narumiya et al. 1999). This pathway is implicated in sex determination via activation and nuclear translocation of SOX-9 (Malki et al. 2005). DP2 is a Gαi-coupled receptor, which enhances intracellular calcium and decreases cAMP leading to the migration of Th2 cells, eosinophils, and basophils (Hirai et al. 2001). PGD2 released by mast cells is also implicated in asthma (Matsuoka et al. 2000) and possess tumor suppressor activities (Wu et al. 2012).
1.3.4 PGF2α
PGF2α is implicated in reproduction by stimulating ovulation, luteolysis, contraction of uterine smooth muscle, and initiation of parturition (Yount and Lassiter 2013; Sugimoto et al. 1997; Saito et al. 2003; De Rensis et al. 2012). PGF2α plays also a role in renal salt transport, myocardial dysfunction, brain damage, and mechanical allodynia (Breyer and Breyer 2001; Kunori et al. 2009; Takayama et al. 2005; Saleem et al. 2009). PGF2α is generated by PGF synthase and mediates its effects through FP receptors. Two variants of FP receptors have been described, FPa and FPb, and both of them are associated with a Gαq protein, which activates phospholipase C and leads to an increase of intracellular Ca+2 and PKC activation (Ito et al. 1994; Bos et al. 2004). PGF2α is produced in human myometrium and ovary (Matsumoto et al. 1997; Sugimoto et al. 1997) and interestingly, its expression is stimulated by oxytocin in endometrial cells (Asselin et al. 1997).
1.3.5 TXA2
TXA2 (thromboxane) produced by TXA2 synthase is a crucial mediator of platelet adhesion and aggregation, smooth muscle constriction, and activation of endothelial inflammatory response that promotes vasoconstriction (Bos et al. 2004; Ricciotti and FitzGerald 2011). TXA2 is preferentially expressed in platelets but can also be expressed in macrophages (Bos et al. 2004; Ricciotti and FitzGerald 2011). Secreted TXA2 mediates its effects through the binding to a membrane Gαq-coupled receptor named “TP receptor”, which is mainly expressed in platelets. Once activated, this Gαq protein activates phospholipase C, which in turn promotes Ca+2 release and PKC activation, leading to platelet aggregation. The production of TXA2 is implicated in several cardiovascular patho-physiological processes including myocardial infarction, atherosclerosis, thrombosis, and stroke (Bos et al. 2004; Ricciotti and FitzGerald 2011).
1.4 Prostaglandin Transporter and 15-Hydroxyprostaglandin Dehydrogenase
The release of prostaglandins can be mediated by simple diffusion across the cell membrane, but recent findings have suggested that specific carriers are necessary for their transport. The prostaglandin transporter (PGT) is a broadly expressed transporter, which possess the ability to uptake various prostaglandins (i.e. PGE2, PGF2α and PGD2) from the extracellular milieu (Kanai et al. 1995; Schuster 2002). Moreover, this uptake is a prerequisite for the inactivation of PGE2 and PGF2α in the cytosol, where 15-hydroxyprostaglandin dehydrogenase (15-PGDH) converts these prostaglandins into their stable 13,14-dihydro-15-keto-PGE2 (PGEM) and 13,14-dihydro-15-keto-PGF2α forms (Holla et al. 2008; Wang and Dubois 2010). Non-enzymatic metabolism is attributed to turnover of other prostaglandins (Wang and DuBois 2007). Interestingly, it has been reported that 15-PGDH is reduced in human colorectal and gastric cancer, thus increasing the amount of PGE2 in the tumor microenvironment and favors immune escape, cancer cell proliferation, and survival (Backlund et al. 2008; Yan et al. 2004).
2 Post-transcriptional Regulation of Prostaglandin Synthesis: Mechanisms of Messenger RNA Decay
Messenger RNA turnover is a highly regulated cellular process that can occur in a rapid, acute manner in response to intracellular and extracellular signals (Garneau et al. 2007). The necessity of post-transcriptional regulation is apparent as 40–50 % of global gene expression changes in response to cellular signals occur at the level of mRNA stability (Balagopal et al. 2012; Fan et al. 2002; Cheadle et al. 2005). Following transcription, eukaryotic mRNAs undergo splicing, RNA editing, and the addition of the 5′ 7-methylguanosine cap and 3′ poly(A) tail (Lutz and Cornett 2013). The 5′ cap and 3′ poly(A) tail, along with other transcript-specific, cis-acting regulatory elements, associate with RNA-binding proteins in order to promote mRNA nuclear export and translation initiation (Day and Tuite 1998; Garneau et al. 2007). Cytoplasmic mRNA decay initiates with the shortening of the poly(A) tail by a complex of enzymes known as mRNA deadenylases, with mammalian cells utilizing three major deadenylation complexes, the Ccr4/Caf1/Not (Caf1) complex, the poly A-specific ribonuclease (PARN ) complex, and the Pan2/Pan3 complex (Sanduja et al. 2012; Chen and Shyu 2011; Fabian et al. 2013; Funakoshi et al. 2007; Goldstrohm and Wickens 2008). At this point, degradation initiating at the 5′ end involves removal of the 5′-7-methyl guanosine cap by the decapping complex Dcp1/Dcp2, leaving the mRNA body susceptible to degradation by the 5′-3′ exonuclease Xrn1 (Garneau et al. 2007; Schoenberg and Maquat 2012; Balagopal et al. 2012). Alternatively, the mRNA can be degraded by 3′-to-5′ exonucleolytic degradation through a complex of exonucleases known as the exosome (Garneau et al. 2007; Balagopal et al. 2012). Many mRNAs targeted for degradation are localized to processing (P)-bodies, which are small cytoplasmic foci that contain components of the 3′-to-5′ and 5′-to-3′ decay machinery along with factors involved in microRNA (miRNA) silencing, nonsense-mediated decay (NMD), and translational silencing (Eulalio et al. 2007; Kulkarni et al. 2010; Zheng et al. 2011; Blanco et al. 2014). Recent evidence demonstrating COX-2 mRNA localization occurring at P-bodies in a signal-dependent manner illustrates the functional significance these discrete cytoplasmic RNA granules have upon prostaglandin biosynthesis (Blanco et al. 2014). The pathways illustrating general cytoplasmic mRNA decay are shown in Fig. 9.2. This section will focus on the functional contribution of regulatory elements within the mRNA 3′ untranslated region (3′UTR ) as well as the role of RNA-binding proteins, miRNAs , and P-bodies upon in prostaglandin biosynthesis.
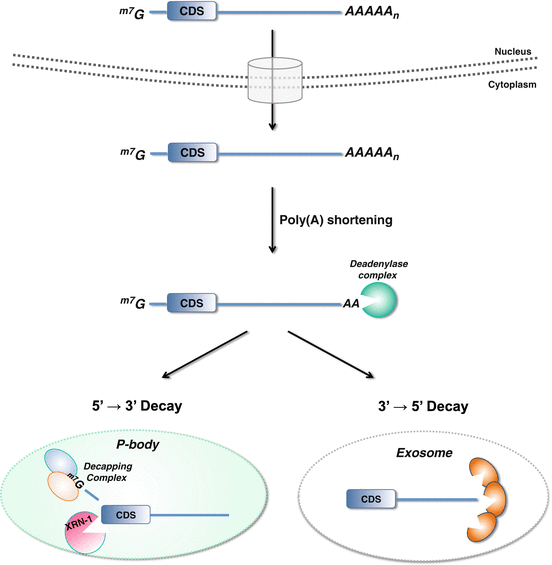
Fig. 9.2
Cytoplasmic mRNA decay pathways. A mRNA transcript undergoes nuclear splicing, and addition of a 5′ 7-methylguanosine cap (m7G) and 3′ poly(A) tail, is exported to the cytoplasm. Degradation initiated through poly(A) shortening by the deadenylase complex is the rate-limiting step of mRNA decay. Once partially deadenylated to 10–15 adenosines, the mRNA transcript can be sequestered to P-bodies wherein mRNA decapping and subsequent Xrn-1-mediated decay occurs. Alternatively, mRNA transcripts may be degraded from the 3′ end by a complex of exonucleases that comprise the exosome
2.1 Post-transcriptional Regulation and AU-Rich 3′UTR Elements
The mRNA 3′UTR is a critical region of the transcript that mediates the interaction with RNA-binding proteins and miRNAs . Considerable evidence exists to support the notion that highly conserved 3′UTR elements play a critical role in post-transcriptional regulation of prostaglandin biosynthesis, with a majority of studies pertaining to regulation of COX-2 expression (Raz et al. 1989; Bailey and Verma 1990; Dixon 2003; Moore et al. 2011).
A commonly observed, well-characterized 3′UTR feature observed in many inflammatory cytokines, growth factors, and proto-oncogenes is the AU-rich element, or ARE (Barreau et al. 2005; Beisang and Bohjanen 2012). The presence of one or several AREs in the mRNA 3′UTR characterizes the short-lived lifespan of many immediate-early response genes and the importance of this particular RNA element is evident, since estimates ranging from 5 to 8 % and 16 % of coding genes contain a 3′UTR ARE sequence (Bakheet et al. 2006; Gruber et al. 2010). The functional ARE is characterized by the 3′UTR consensus sequence AUUUA and is most often composed of multiple AUUUA motif copies. AREs are organized into several classes and clusters on the basis of the number and context of the AUUUA pentamer. For example, various immediate-early response genes (e.g. proto-oncogenes) have scattered repeats of the AUUUA motif, whereas mRNAs encoding inflammatory mediators and cytokines (e.g. COX-2) that have multiple repeats of the AUUUA pentamer clustered together (Beisang and Bohjanen 2012; Bakheet et al. 2006). The use of search engines such as AREsite (http://rna.tbi.univie.ac.at/cgi-bin/AREsite.cgi) and ARED (ARE Database; http://brp.kfshrc.edu.sa/ARED/) have provided investigators online tools to identify the presence of AREs in eukaryotic mRNAs (Bakheet et al. 2006; Gruber et al. 2010), and the presence of this 3′UTR element was observed in a majority of PGE2 pathway genes (Moore et al. 2011). The role of ARE-mediated post-transcriptional regulation in prostaglandin biosynthesis has been best described for COX-2 and cPLA2 (Dixon 2004; Young and Dixon 2010; Tay et al. 1994). The functional ARE of COX-2 is comprised of an evolutionarily-conserved 116 nucleotide stretch containing six AUUUA clusters located proximal to the stop codon (Dixon et al. 2000). The COX-2 ARE mediates important interaction with ARE RNA-binding proteins that can either target the mRNA for rapid decay or enhance the half-life of the transcript, depending on the cellular context (Sengupta et al. 2003; Young et al. 2009). Similar to COX-2, AREs present in the cPLA2 exert an mRNA-destabilizing effect and impact overall PGE2 synthesis (Tay et al. 1994). A further refinement of our understanding of these post-transcriptional mechanisms may provide a novel therapeutic window for the development of innovative NSAIDs targeting aberrant ARE function for the treatment of pain, inflammation, and cancer.
2.2 ARE RNA-Binding Proteins
Through its presence, the ARE serves to target mRNAs for rapid degradation and/or translational suppression within the cytoplasm (Barreau et al. 2005; Beisang and Bohjanen 2012). AREs mediate this through association with RNA-binding proteins and miRNAs that bind with high affinity (Beisang and Bohjanen 2012; Stumpo et al. 2010; Young et al. 2012; Jing et al. 2005; Garneau et al. 2007). There are >20 known ARE-binding proteins with a majority being associated with promoting mRNA stabilization , mRNA decay , or controlling translation by directing ARE-containing mRNAs to P-bodies and stress granules (Beisang and Bohjanen 2012; Garneau et al. 2007). Through these mechanisms, ARE-binding proteins exhibit pleiotropic effects on gene expression, since a single ARE-binding protein can bind to multiple mRNAs and binding can occur among different classes of AREs (Lopez de Silanes et al. 2007).
Considerable evidence exists to support the integral role of trans-acting ARE -binding proteins in prostaglandin biosynthesis that allow for fine control of gene expression in response to various cellular cues and signaling within the microenvironment. Various cytoplasmic proteins have been detected to bind AREs and a majority of work has focused on COX-2 ARE-binding proteins with 16 different RNA-binding proteins identified to bind the COX-2 3′UTR (Young and Dixon 2010). This section will describe the most well characterized ARE RNA-binding proteins that regulate COX-2 expression and associated prostaglandin biosynthesis (Fig. 9.3).
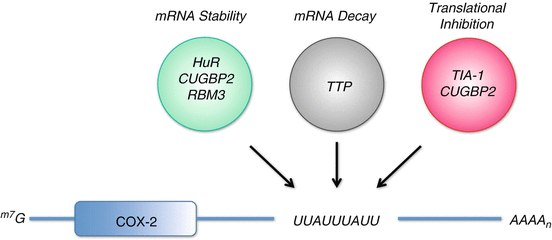
Fig. 9.3
ARE RNA-binding proteins mediating post-transcriptional regulation of COX-2. COX-2 expression is tightly regulated at the post-transcriptional level by RNA-binding proteins that promote mRNA stability (HuR , CUGBP2 , and RBM3), mRNA decay (TTP ), and translational inhibition (TIA-1 and CUGBP2) through their binding of the COX-2 AU-rich element (ARE)
2.2.1 HuR
The HuR protein (ELAVL1, Hu antigen R) is a ubiquitously expressed member of the ELAV (Embryonic-Lethal Abnormal Vision in Drosophila) family of RNA-binding proteins that consists of HuR and primarily neuronal-specific HuB, HuC, and HuD (Brennan and Steitz 2001; Hinman and Lou 2008). It is primarily nuclear localized (>90 %), where it assists in pre-mRNA splicing and cytoplasmic export of mRNA transcripts, followed by rapid nuclear re-localization (Brennan and Steitz 2001; Fan and Steitz 1998; Srikantan and Gorospe 2011). HuR is comprised of two tandemly arrayed RNA-recognition motifs (RRM), followed by a hinge region and a third RRM. The hinge region contains an HNS domain, which mediates nucleo-cytoplasmic shuttling of the HuR protein (Fan and Steitz 1998; Keene 1999).
The ability of HuR to function as an ARE -stability factor and enhance mRNA half-life appears to be linked to its subcellular localization (Brennan and Steitz 2001; Srikantan and Gorospe 2012). In response to cellular stress (e.g., inflammation, hypoxia , DNA damage), HuR translocates to the cytoplasm and enhances the stability of inflammation- and other survival-associated transcripts. Similarly, HuR overexpression observed in a variety of tumor types results in cytoplasmic localization and subsequent ARE-mRNA stabilization (Wang et al. 2013a; Abdelmohsen and Gorospe 2010; Srikantan and Gorospe 2012). Although the precise molecular underpinnings that regulate HuR nucleo-cytoplasmic localization are not entirely known, a variety of cellular signals known to activate MAPK pathways (ERK and p38 kinases), PI-3-kinase pathway, and Wnt signaling pathway, have been shown to trigger HuR cytoplasmic localization and influence ARE-mRNA stabilization (Winzen et al. 1999; Yang et al. 2004; Tran et al. 2003; Ming et al. 2001; Briata et al. 2003).
HuR has been shown to bind and post-transcriptionally regulate numerous ARE -containing transcripts associated with cancer traits and inflammation (Wang et al. 2013a; Abdelmohsen and Gorospe 2010). Based on its ability to bind the COX-2 ARE region consisting of a highly conserved cluster of six AUUUA elements located near the stop codon, HuR has been identified as a trans-acting factor involved in regulating COX-2 expression (Dixon et al. 2001). The enhanced stabilization of COX-2 mRNA observed in colon cancer cells and inflamed myeloid leukocytes is, in part, due to elevated cytoplasmic levels of HuR (Dixon et al. 2001, 2006; Young et al. 2009). Furthermore, several studies indicate that HuR overexpression and cytoplasmic localization is a marker for elevated COX-2 that is correlated with advanced tumor stage and poor clinical outcome (Dixon 2003; Abdelmohsen and Gorospe 2010; Wang et al. 2013a). PLA2 and mPGES-1 have also been identified as a direct targets of HuR, with inflammatory signaling enhancing the association of cPLA2α mRNA with cytosolic HuR (Liao et al. 2011; Lopez de Silanes et al. 2004). These findings suggest HuR to play a distinct regulatory role by integrating expression of factors involved in prostaglandin synthesis through mRNA stabilization .
It is generally accepted that HuR stabilizes mRNAs by competing or displacing destabilizing factors from the ARE (Benjamin and Moroni 2007). Another mechanism indicates that HuR can inhibit mRNA association with the decay machinery or possibly protect the poly(A) tail from degradation (Linker et al. 2005; Lal et al. 2004; Ma et al. 1997). Current evidence now indicates that HuR’s ability to influence stabilization of bound mRNAs can be mediated through HuR’s interplay with miRNAs that associate with the same transcript (Srikantan et al. 2012), and recent findings demonstrate that HuR promotes COX-2 mRNA stabilization by outcompeting miRNAs that share the same ARE binding sites in the COX-2 3′UTR (Young et al. 2012). In line with these findings, HuR has been shown to rescue mRNA transcripts held translationally silenced by miRNAs in P-bodies to promote their translation (Bhattacharyya et al. 2006; Srikantan et al. 2012).
High-throughput based biochemical screens have identified small-molecule compounds with the ability to disrupt HuR /ARE interactions (Meisner and Filipowicz 2010; Meisner et al. 2007; Chae et al. 2009; D’Agostino et al. 2013). Work conducted by Meisner and coworkers had identified and characterized three compounds (okicenone, dehydromutacin, and MS-444) as specific, low-molecular-weight HuR inhibitors (Meisner et al. 2007). These compounds are polyketides purified from plant and microbial extracts and the latter, MS-444, has been extensively studied with regard to its mechanism of action and ability to inhibit HuR. Mechanistically, MS-444 inhibits HuR homodimerization and blocks its cytoplasmic export. The impact of this results in loss of HuR-dependent mRNA stabilization and disruption of HuR/miRNA interaction (Young et al. 2012; Meisner et al. 2007).
2.2.2 TTP
Tristetraprolin (TTP , ZFP36, TIS11, NUP475) is a member of a small family of Cys3His zinc finger proteins. This family is comprised of TTP, ZFP36L1 (BRF-1), and ZFP36L2 (BRF-2), all which have been shown to play a critical role in regulated mRNA decay (Sanduja et al. 2011; Blackshear 2002; Ciais et al. 2013). TTP is one of the best-characterized post-transcriptional regulators, whereby it’s binding of AREs promotes rapid mRNA decay (Brooks and Blackshear 2013; Carballo et al. 1998). The binding of TTP to AREs targets the transcript for rapid degradation through the recruitment of mRNA deadenylases, translational repressors, and mRNA decapping proteins onto the mRNA transcript (Chen et al. 2001; Mukherjee et al. 2002; Fenger-Gron et al. 2005; Lykke-Andersen and Wagner 2005; Franks and Lykke-Andersen 2007; Hau et al. 2007). At this point, TTP promotes the nucleation of P-bodies by delivering its cargo mRNA for subsequent degradation (Kedersha et al. 2005; Blanco et al. 2014; Franks and Lykke-Andersen 2007) (Fig. 9.4a). TTP has also been implicated as a novel mediator of miRNA-dependent post-transcriptional regulation through its ability to associate with the argonaute (Ago) protein family members and promote decay of ARE -containing mRNAs localized to P-bodies (Eulalio et al. 2007; Jing et al. 2005). Alternatively, TTP can promote exosome-mediated mRNA degradation through its ability to interact with the exosome and recruit it to the AU-rich mRNAs (Chen et al. 2001).
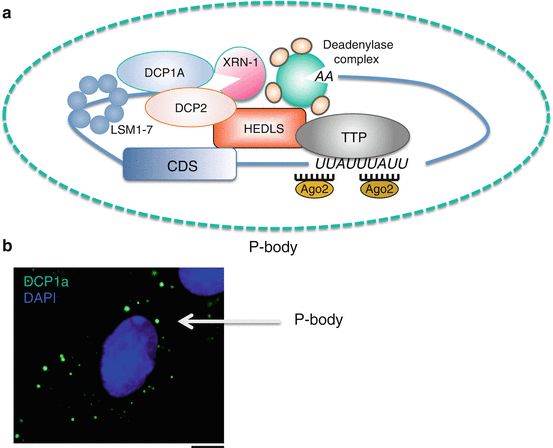
Fig. 9.4
P-bodies are sites of mRNA storage or decay. (a) Schematic of a P-body. Once an ARE -containing mRNA is targeted for decay and partially deadenylated, TTP recruits mRNA degradation machinery facilitating mRNA decapping (Dcp1a/Dcp2/Hedls/Lsm1-7) and further deadenylation . RNA degradation occurs in a 5′→3′ direction by the exonuclease Xrn-1. TTP also facilitates miRNA-mediated mRNA degradation through association with Ago proteins. (b) Example of P-bodies present in intestinal epithelial cells. P-bodies were detected by immunofluorescence staining for the P-body marker DCP1a (green signal). Cellular nuclei are visualized by DAPI (blue signal). Scale bar 10 μm
TTP plays a critical role in normal physiology as TTP knockout mice develop early on a severe syndrome of growth retardation, cachexia, arthritis, inflammation and autoimmunity (Taylor et al. 1996). Based on this severe inflammatory phenotype, inflammatory mediators such as TNF-α, GM-CSF and COX-2 have been identified as ARE -containing targets of TTP (Lai et al. 1999; Carballo et al. 2000; Brooks and Blackshear 2013; Sawaoka et al. 2003; Young et al. 2009). TTP preferentially binds the nonameric sequence motif, UUAUUUAUU, the core destabilizing element of many ARE-containing mRNAs (Blackshear et al. 2003; Brewer et al. 2004). As nonameric sequences are the second most abundant sequence motif present in genes involved in the PGE2 pathway (Moore et al. 2011), TTP’s role in the regulation of prostaglandin biosynthesis may extend beyond COX-2 regulation.
TTP is an inducible, immediate-early response gene whose expression can be induced by mitogenic stimuli such as insulin , growth factors (e.g., TGF-β), and pro-inflammatory signals (Blanco et al. 2014; Ogawa et al. 2003). However, a consistent loss of TTP expression occurs in a variety of human cancers such as breast, colon, cervix, prostate, and lung (Sanduja et al. 2009, 2012; Brennan et al. 2009; Young et al. 2009), and suppressed TTP expression can serve as a negative prognostic breast cancer indicator (Brennan et al. 2009). These findings indicate that the presence of TTP in normal tissues serves a protective role by controlling expression of various pro-inflammatory and prostaglandin synthesis mediators, while loss of TTP expression in tumors contributes to aberrant overexpression of these transcripts.
2.2.3 RBM3
RBM3 (RNA-binding motif protein 3) is a member of a family of glycine-rich RNA-binding proteins (Derry et al. 1995). RBM3 is comprised of a single RRM and has been shown to regulate COX-2 expression through ARE binding (Cok and Morrison 2001; Sureban et al. 2008). In addition to COX-2, RBM3 has been shown to bind and stabilize the IL-8 and VEGF mRNAs and prevents mitotic catastrophe in colon cancer cells (Sureban et al. 2008). Together with the findings that RBM3 is significantly upregulated in colorectal tumors and overexpression promotes oncogenic transformation in fibroblasts (Sureban et al. 2008), identify a pro-oncogenic role of this ARE-binding protein.
Previously, yeast two-hybrid screens identified the ability of RBM3 to interact with HuR (Anant et al. 2010). Although the significance of this interaction remains to be determined, RBM3, like HuR, is a nucleo-cytoplasmic shuttling protein whose cytoplasmic export is triggered by stresses such as hypothermia, hypoxia , serum deprivation (Wellmann et al. 2010). Through its ability to promote the translation of otherwise unstable mRNA transcripts, RBM3 is being recognized as a contributing factor promoting enhanced prostaglandin levels in pathogenic states.
2.2.4 CUGBP2
A member of the CUGBP-ETR-3-like factors family, CUG triplet repeat-binding protein 2 (CUGBP2 ) is a ubiquitously expressed RNA-binding protein comprised of two N-terminal RRMs and one C-terminal RRM (Mukhopadhyay et al. 2003; Murmu et al. 2004).
Like other members of this family of proteins, CUGBP2 regulates alternative splicing , RNA editing, and mRNA translation (Anant et al. 2001). Importantly, CUGBP2 displays high affinity for the COX-2 ARE and has been shown to regulate its expression in response to ionizing radiation in colon cancer cells and growth factor stimulation of colonic stromal cells (Mukhopadhyay et al. 2003; Walker et al. 2010). Studies in vascular smooth muscle cells demonstrated that the association between CUGBP2 and the COX-2 ARE occurs in response to platelet-derived growth factor (PDGF)-dependent phosphorylation of the CUGBP2 protein. To this extent, the kinases c-Src and c-Abl directly phosphorylate CUGBP2 thus mediating its ability to inhibit ribosomal loading of the COX-2 mRNA (Xu et al. 2007). In a model of cardiac hypertrophy, CUGBP2 was identified to control pro-inflammatory stimulus involving COX-2/PGE2 and promote subcellular mRNA trafficking specific cytoplasmic stress granules to maintain homeostasis in cardiac cells (Moraes et al. 2013).
CUGBP2 exhibits a complex biology such that it promotes COX-2 mRNA stabilization coupled with translational inhibition. Although CUGBP2 and HuR share similar affinities for the same binding sites in the COX-2 ARE , CUGBP2 effectively outcompetes HuR and promotes translational silencing of COX-2 (Murmu et al. 2004). This dynamic, seemingly antagonistic role of CUGBP2 indicates a possible role for this protein in the early stages of inflammation-induced tumorigenesis by attenuating PGE2 synthesis through repression of COX-2 expression.
2.2.5 TIA-1
T cell intracellular antigen 1 (TIA-1) is an RNA-binding protein originally identified in activated T lymphocytes and is comprised of three RRMs. TIA-1 displays high affinity for short repeats of uridylate rich regions in non-coding regions (e.g., 3′UTR ) of many pro-inflammatory cytokines (Tian et al. 1991; Lopez de Silanes et al. 2005). TIA-1 acts as a translational repressor, and under conditions of stress , it sequesters its target mRNA transcripts to cytoplasmic RNA granules known as stress granules (SGs). There, mRNAs are held translationally silenced and are sorted to re-enter translation or proceed to ARE -mediated mRNA decay in P-bodies (Kedersha et al. 2005; Anderson and Kedersha 2008). Along with it recognized role in translational suppression , TIA-1 is also implicated in alternative splicing regulation of various pre-mRNAs through binding adjacent to exon/intron boundaries (Wang et al. 2010b, 2014).
TIA-1 has been shown to bind the COX-2 ARE and regulate its expression through translational inhibition without altering COX-2 mRNA turnover. However, TIA-1-mediated regulation of COX-2 is deficient in colon cancer cells, thus contributing to increased polysome association with the COX-2 mRNA (Dixon et al. 2003). This regulation of COX-2 expression was also observed in TIA-1-deficient fibroblasts that produce significantly more COX-2 protein and PGE2 than wild-type fibroblasts. The physiological role of TIA-1 was best described in studies with the TIA-1 knockout mice. Similar to TTP deficient mice, TIA-1 null mice exhibited aberrant expression of pro-inflammatory cytokines (e.g., COX-2), thus leading to the development of arthritis (Phillips et al. 2004). These findings indicate that loss of TIA-1-mediated regulation of COX-2 contributes to increase COX-2 protein expression and PGE2 synthesis in cancer and inflammation.
2.3 MicroRNAs
MicroRNAs (miRNAs ) represent a class of endogenous small non-coding RNAs involved in the post-transcriptional regulation of broad biological functions (Frankel and Lund 2012; Ambros 2004; Graves and Zeng 2012). MiRNAs are ≈22 nucleotides and regulate the expression of complementary target mRNA(s) primarily through 3′UTR binding by influencing translation and by causing degradation of target mRNAs (Fabian et al. 2010) and findings have implicated miRNA-mediated mRNA decay to be the predominant mechanism (Guo et al. 2010). MiRNAs are transcribed by RNA polymerase II to produce a longer primary precursor pri-miRNA (Graves and Zeng 2012). This pri-miRNA is maturated by the Drosha endonuclease microprocessor complex to release ≈60–70 nucleotides long stem-loop intermediate pre-miRNAs. Alternatively, the pre-miRNA can be generated in a Drosha-independent manner, through intronic pri-miRNAs, which are maturated by the spliceosome into pre-miRNAs (Bartel 2004). The pre-miRNA is then exported in the cytosol through by exportin 5 and processed by Dicer to release a ≈22 nucleotides mature miRNA (guide strand)/miRNA* (passenger strand) duplex (Bartel 2004). According to the current canonical model, the guide strand is associated with argonaute (Ago) proteins and incorporated in the RNA-Inducing Silencing Complex (RISC ) where the miRNA binds its mRNA target(s), while the miR* is degraded (Yates et al. 2013; Bartel 2004). Current findings indicate that the miR* can also play an important role in biological processes by targeting different mRNAs than their respective guide strands (e.g. miR-21* in kidney fibrosis or miR-199* in PGs biosynthesis) (Rayner et al. 2011). Consequently, the role of the passenger strand needs to be considered with caution, especially in studies using knockout mice, where the guide and the passenger strands are both deleted.
miRNAs regulate more than 60 % of coding mRNAs and their biological effects are organized in a complex network (Friedman et al. 2009). This level of complexity occurs through the pleiotropic actions of miRNAs targeting numerous mRNAs and it is likely that the phenotype arising alteration of one particular miRNA is the consequence of a concerted action on several targets (Ambros 2004; Bartel 2004). A further degree of complexity is provided by other non-coding RNAs such as competitive endogenous RNAs (ceRNA including pseudogenes, circular RNAs or long non-coding RNAs), which are potent regulators of miRNAs by acting as a “sink” for miRNAs (Tay et al. 2014). Other post-transcriptional regulators need also to be taken into account in the miRNAs network, such as RNA-binding proteins including ARE -binding proteins, which can promote or inhibit miRNAs function (Adams et al. 2014; Young et al. 2012; Srikantan et al. 2012).
miRNAs have been associated with a variety of pathologies including metabolic diseases, cardiovascular disease, and cancer (Farazi et al. 2013). Alterations in expression or activity of miRNAs regulating the expression of key enzymes involved in prostaglandin homeostasis are likely to contribute to the onset and progression of these pathologies. Numerous miRNAs have been predicted by different computational methods to bind directly to the 3′UTR of key enzymes involved in prostaglandin biosynthesis (e.g., TargetScan, miRanda, microRNA .org) (Moore et al. 2011). Several miRNAs targeting phospholipase A2, phospholipase C, COX-1, COX-2, as well as prostaglandin synthases have been identified, with a majority of efforts on investigating COX-2 regulation (Moore et al. 2011; Young and Dixon 2010). Furthermore, miRNAs can affect the expression of these enzymes through indirect mechanisms, for instance targeting transcription factors controlling the expression of the enzymes implicated in PG synthesis (e.g. NFκB) (Ma et al. 2011). Herein, we will primarily discuss functionally validated miRNAs regulating the expression of the different enzymes involved in PGs synthesis (Table 9.1).
Table 9.1
Validated microRNAs targeting genes involved in prostaglandin synthesis
Gene | Validated miRNAsa (References) |
|---|---|
cPLA2-α (PLA2G4A) cPLA2-β (PLA2G4B) | N.D. miR-338 (Montenegro et al. 2009) |
Phospholipase C (PLCγ1) | miR-200b, miR-200c, miR-429 (Uhlmann et al. 2010) miR-218-2 (Guan et al. 2013) |
COX-1 (PTGS1) | N.D. |
COX-2 (PTGS2) | miR-101a (Chakrabarty et al. 2007; Daikoku et al. 2008; Strillacci et al. 2009; Tanaka et al. 2009; He et al. 2012; Wang et al. 2010a) miR-137 (Chen et al. 2012) miR-199a-3p/miR-214 (Williams et al. 2012) miR-542-3p (Moore et al. 2012) |
mPGES-1 (PTGES) mPGES-2 (PTGES2) cPGES (PTGES3) | N.D. miR-146a (Matysiak et al. 2013) N.D. |
EP1 Receptor (PTGER1) EP2 Receptor (PTGER2) EP3 Receptor (PTGER3) EP4 Receptor (PTGER4) | N.D. N.D. N.D. miR-101 (Chandramouli et al. 2012) |
Prostacyclin synthase (PTGIS) | N.D. |
IP Receptor (PTGIR) | N.D. |
PGD2 synthase (PTGDS) PGD2 synthase (HPGDS) | N.D. N.D. |
DP Receptor (PTGDR) DP Receptor (PTGDR2) | N.D. N.D. |
PGF2α Synthase (PGFS) | N.D. |
FP Receptor (PTGFR) | N.D. |
TXA2 Synthase (TBXAS1) | N.D. |
TXA2 Receptor (TBXA2R) | miR-765 (Nossent et al. 2011) |
Prostaglandin Transporter (SLCO2A1, PGT) | N.D. |
15-Hydroxyprostaglandin Dehydrogenase (HPGD, 15-PGDH) | miR-21 (Lu et al. 2014) |
2.3.1 miR-16-1 and miR-15a
The first reports implicating a role for miRNAs in cancer progression examined miR-16-1/15a cluster and its function in chronic lymphocytic leukemia (CLL) due to a commonly deleted genomic region in CLL accompanied with loss of miR-16-1 and miR-15a expression (Calin et al. 2002; Dohner et al. 2000). The expression of miR-16 is also reduced in other cancers, including colorectal, liver, prostate, lung, and gliomas (Young et al. 2012; Ge et al. 2014; Yang et al. 2014; Bottoni et al. 2005; Lu et al. 2005; Bonci et al. 2008; Navarro et al. 2011). Various mechanisms are responsible for the downregulation of miR-15a and miR-16 in cancers. MiR-16 is encoded within an intronic region of DLEU2 (deleted in lymphocytic leukemia) on chromosome 13q14, a region which is frequently deleted in CLL (Pekarsky and Croce 2014). Other evidence indicates downregulation of miR-16 can be also mediated by epigenetic events such as HDACs overexpression (Sampath et al. 2012). MiR-16-1/15a serves in a tumor suppressor capacity by regulating expression of anti-apoptotic factors such as Mcl-1 and Bcl-2 (Yang et al. 2014; Liu et al. 2014; Sanchez-Beato et al. 2003; Cimmino et al. 2005). MiR-16 in particular has additionally been shown to play a role in cell cycle maintenance through regulation of several cell cycle regulatory genes (Liu et al. 2008).
The initial connection between miR-16 and COX-2 was demonstrated in the work by Jing et al. that identified the ability of miR-16 to promote degradation of ARE -containing transcripts as miR-16-1 contains sequence homology to AU-rich elements (Jing et al. 2005). Other studies have validated miR-16-1 targeting of the COX-2 3′UTR in work examining COX-2 3′UTR-mediated post-transcriptional regulation by miR-16-1 in response to diabetic stimuli in leukocytes (Shanmugam et al. 2008). A similar link between miR-16 and COX-2 was observed in hepatoma and colorectal cancer cells (Agra Andrieu et al. 2012; Young et al. 2012). While these findings underscore the importance of miR-16 in PG synthesis, RNA-binding proteins also regulate miR-16 function. MiR-16-induced mRNA decay requires the mRNA-destabilizing factor TTP to promote decay of ARE-mRNAs (Jing et al. 2005). Other proteins able to stabilize COX-2 mRNA, such as HuR , can interfere with miR-16 binding of the COX-2 ARE by binding miR-16 (Young et al. 2012). Similarly, hnRNPK (heterogeneous nuclear ribonucleoprotein K) leads to a decreased binding of miR-16 to COX-2 mRNA (Shanmugam et al. 2008). Interestingly, studies have shown miR-16 also directly regulates HuR expression. In breast cancer, miR-16-1 has been implicated to repress translation of HuR and loss of miR-16-1 was correlated with HuR overexpression (Xu et al. 2010). Consistent with this, miR-16 has been shown to decrease COX-2 expression in hepatocellular carcinoma by two distinct mechanisms. First, through direct binding of the COX-2 3′UTR and impeding translation and second, miR-16 downregulates HuR expression (Agra Andrieu et al. 2012).
2.3.2 miR-200 Family
The miR-200 family encompasses five miRNAs , including miR-200a, miR-200b, miR-200d, miR-141 and miR-429, which are encoded by two different polycistronic pri-miRNA transcripts (Moore et al. 2011). MiR-200a, b and miR-429 are located on Chr-1, while miR-200c and miR-141 are on Chr-12 (Gregory et al. 2008). Interestingly, the chromosomal regions encoding miR-200 family members are frequently deleted in cancers, leading to a downregulation of these miRNA (Mongroo and Rustgi 2010). MiR-200 family is also downregulated in non-cancerous diseases such as liver fibrosis (Murakami et al. 2011). The link between miR-200 family and PGs synthesis was highlighted in breast cancer cells, where overexpression of miR-200b, miR-200c and miR-429 promoted downregulation of PLCγ1 mRNA expression, and an increase of PLCγ1 mRNA was observed with a miR-200bc/429 inhibitor in MCF7 cells (Uhlmann et al. 2010). Moreover, the increase of miR-200bc/429 in breast cancer cells reduces cell viability and induces apoptosis (Uhlmann et al. 2010).
2.3.3 miR-137
MiR-137 loss has been linked with several diseases such as schizophrenia (Schmidt et al. 2013) and cancers including lung, ovarian, and neuroblastoma (Li et al. 2014; Guo et al. 2013; Althoff et al. 2013). Based on predicted binding sites for miR-137 in the COX-2 3′UTR , expression of miR-137 in glioma cells promoted COX-2 downregulation and inhibition of cell proliferation and invasion. These observations are coupled with an inverse correlation between the expression of miR-137 and COX-2 in glioblastoma cells, suggesting a potential role of miR-137 as a tumor suppressor in glioblastoma (Chen et al. 2012).
2.3.4 miR-21
MiR-21 is the only miRNA known to date to be upregulated in all human cancers (Pan et al. 2011; Volinia et al. 2006). A large amount of experimental data has demonstrated that overexpression of miR-21 impacts various aspects of tumorigenesis including tumor cell proliferation, evasion of apoptosis, and invasion and metastasis (Pan et al. 2011). In addition, miR-21 is implicated in metabolic disorders such as NAFLD (Non-Alcoholic Fatty Liver Disease), where its ability to downregulate PTEN affects insulin sensitivity (Vinciguerra et al. 2009; Ling et al. 2012).
MiR-21 is an important regulator of prostaglandin synthesis by directly targeting 15-hydroxyprostaglandin dehydrogenase (15-PGDH), which converts PGE2 into its inactive form, 15-keto-PGE2 (Lu et al. 2014). In cholangiocarcinoma, where elevated expression of miR-21 is correlated with enhanced PGE2 levels and loss of 15-PGDH (Lu et al. 2014). It has been found that nicotine stimulation upregulates the expression of miR-21 in gastric cancer cells and this effect was in an NFκB-dependent manner (Shin et al. 2011), indicating that ability of miR-21 to affect PGs synthesis can be also influenced by indirect effects. On these same lines, a connection between PTEN loss and the increase of cPLA2 has been found in the prostate cancer cells (Vignarajan et al. 2014). Thus, the ability of miR-21 to downregulate PTEN may indirectly affect cPLA2 expression and impact PGs biosynthesis.
2.3.5 miR-101
The genomic locus encoding miR-101 is located on Chr 1p31.3 and loss of miR-101 has been observed in various tumor types including in 67 % of metastatic cancer cells, suggesting its involvement in tumor cell invasion (Varambally et al. 2008; Su et al. 2009; Liang et al. 2014; Hiroki et al. 2010; Zhang et al. 2011; Strillacci et al. 2009). In endometrial cancer, miR-101 is involved in regulating expression of oncogenes such as Mcl-1 and Fos (Konno et al. 2014). MiR-101 has been shown to regulate COX-2 expression in a number of models. Using a toxin-induced liver injury, it has been shown that miR-101 regulates COX-2 expression and prostaglandin synthesis (Yoshioka et al. 2011). Another study has also shown the downregulation of miR-101 and a concomitant upregulation of COX-2 in cervical cancer tissue from patients, indicating miR-101 loss as a biomarker of COX-2 elevation (Lin et al. 2014). Supporting these findings, it has been found that the expression of miR-101 is low in HeLa cells and the restoration of miR-101 expression downregulates COX-2 expression with decreased cell proliferation and migration and an induction of apoptosis (Huang et al. 2013). Similar results were observed in colorectal and gastric cancer cells where COX-2 downregulation occurred following miR-101 overexpression, where miR-101 was observed to interact with COX-2 3′UTR and reduce protein expression (Hao et al. 2011; Wang et al. 2010a; Strillacci et al. 2009). miR-101 also plays an important role in embryo implantation by targeting directly COX-2. It has been shown that miR-101 is induced in mouse uterus during embryo implantation, while the expression of COX-2 is reduced (Chakrabarty et al. 2007). Given the importance of COX-2-derived prostaglandins during embryo implantation (Kennedy et al. 2007; Lim et al. 1999), the ability of miR-101 to downregulate COX-2 expression may influence the implantation process.
Beside its ability to downregulate COX-2, it has been suggested that miR-101 downregulates also EP4 receptor expression. An inverse correlation between the expression of miR-101 and EP4 has been observed in colorectal cancer and the overexpression of miR-101 in colon cancer cells causes a reduction of EP4 expression and impairs cell proliferation and migration (Chandramouli et al. 2012). These findings show that miR-101 is a potent inhibitor of PGE2 signaling by impacting PGE2 synthesis and its transduction pathway.
2.3.6 miR-146a
This miRNA is induced by NFκB and is an important regulator of innate immune response by targeting directly mRNAs in the TNFα and IL-6 signaling pathways (Taganov et al. 2006). More recently, miR-146a was found to be an important regulator of PG synthesis. The link was shown in fibroblasts from chronic obstructive pulmonary disease patients (COPD) where increases COX-2/PGE2 expression were observed (Sato et al. 2010). This effect was partially associated with reduced levels of miR-146a and inhibition of COX-2 mRNA degradation , and overexpression of miR146a prevented IL-1β or TNFα-induced COX-2 expression and PGE2 synthesis (Sato et al. 2010). Interestingly, the expression of miR-146a is positively regulated by NFκB, thus suggesting that the induction of this miRNA may represent part of a negative feedback loop aiming at lowering PG synthesis (Ghose et al. 2011; Taganov et al. 2006).
Recent findings have shown that miR-146a can also affect other factors involved in PG synthesis. MiR-146a inhibits an immunoregulatory function of bone marrow stem cells (BMSCs) by targeting directly mPGES-2 mRNA and limiting PGE2 synthesis (Matysiak et al. 2013). In this study, neuronal differentiated BMSC (nBMSC) express a higher level of miR-146a compared to non-differentiated BMSCs and inhibition of miR-146a with a selective antagomiR in nBMSC leads to an increase of PGE2 synthesis (Matysiak et al. 2013). Alteration of miR-146a expression has been observed in a variety of pathologies such as lung cancer (Cornett and Lutz 2014), where the expression of miR-146a is lost. Paradoxically, herpes simplex virus-1 can induce the expression of miR-146a in neuronal cells, which leads to an induction of inflammation and particularly an induction of PLA2 and COX-2 (Hill et al. 2009). These findings suggest that different mechanisms may occur, depending on the disease etiology and the cellular context.
2.3.7 miR-26b
MiR-26b is encoded within the intronic region of genes encoding for proteins of carboxy-terminal domain RNA polymerase II polypeptide A small phosphatase (CTDSP) family (Zhu et al. 2012). The expression of this miRNA is reduced in different neoplasias such as hepatocellular, nasopharyngeal, lung, and breast cancers (Shen et al. 2014; Ji et al. 2010; Gao et al. 2011; Li et al. 2013). MiR-26b is associated with obesity and its expression is downregulated by free fatty acids, glucose, or glucocorticoids in adipocytes, indicating a link between this miRNA and insulin resistance (Xu et al. 2014). It has been shown that miR-26b regulates COX-2 expression in desferrioxamine-treated nasopharyngeal cancer cells and overexpression of miR-26b leads to a reduction in cell proliferation associated with the COX-2 downregulation (Ji et al. 2010). In breast cancer, the expression of miR-26b contributes to decreased COX-2 mRNA and protein levels and inhibits cell proliferation (Li et al. 2013).
2.3.8 miR-199/miR-199*
Currently, few studies have documented the function of miR-199 in physiological processes but its overexpression has been associated with liver fibrosis (Murakami et al. 2011). miR-199 appears to play an important function during pregnancy given that its downregulation is observed in laboring myometrium while COX-2 expression is increased (Williams et al. 2012). More strikingly, overexpression of miR-199-3p/miR-214 using miRNA mimics in myometrial cells reduced COX-2 protein expression (Williams et al. 2012). The passenger strand of miR-199, miR-199* is also a potent inhibitor of COX-2 expression. In human osteoarthritis chondrocytes, it has been shown that miR-199* inhibits IL-1β-induced COX-2 expression (Akhtar and Haqqi 2012). Interestingly, IL-1β-induced p38 MAPK activation is inversely correlated with the level of miR-199* and an induction of miR-199* was observed in chondrocytes treated with a p38 inhibitor (Akhtar and Haqqi 2012). COX-2-derived prostaglandins play an important role during embryo implantation (Kennedy et al. 2007; Lim et al. 1999). The role of miRNA in this process has been observed with the expression of miR-101 and miR-199* increased in the murine uterus during embryo implantation, while the expression of COX-2 is reduced (Chakrabarty et al. 2007). Consistent with these findings, the expression of miR-199a* are reduced in a model of endometrial cancer induced by PTEN loss, while the expression of COX-2 is induced (Daikoku et al. 2008).
2.3.9 miR-143/145
The miR-143/145 cluster is located on Chr 5q32 and current findings suggest that they originate from the same primary miRNA (Moore et al. 2011). Downregulation of miR-143/145 is observed in several disorders including colorectal, prostate, esophageal, bladder, osteosarcoma cancers, and hematologic malignancies such as B cell lymphoma or chronic lymphocytic leukemia (Luo et al. 2011; Kojima et al. 2014; Wu et al. 2011; Yoshino et al. 2013; Hu et al. 2012; Fabbri et al. 2008). The suppressive effect of miR-143 on COX-2 expression has been described in gastric, bladder, and pancreatic cancer cells through targeting the COX-2 3′UTR (Wu et al. 2013; Song et al. 2011; Pham et al. 2013). In addition, miR-143-3p and miR-143-5p decrease cell viability and proliferation and induce apoptosis in gastric cancer cells (Wu et al. 2013). MiR-143 expression is diminished in bladder cancer cells and its overexpression leads to COX-2 downregulation and suppression of metastasis (Song et al. 2011).
2.3.10 miR-542-3p
Very few studies have been performed on this miRNAs but current findings suggest that miR-542 acts as a tumor suppressor, through its ability to downregulate the pro-survival oncogene survivin (Althoff et al. 2014). More recently, it has been shown that miR-542-3p regulates PG synthesis by directly targeting the COX-2 3′UTR (Moore et al. 2012). Interestingly, the 3′UTR region targeted by miR-542-3p contains a common single nucleotide polymorphism (SNP) in the COX-2 gene at position 8473 in exon 10 (T8473C; rs5275) that is associated with increased risk and/or NSAID responsiveness in a number of cancers where COX-2 over-expression is a contributing factor. Mir-542-3p was identified to bind transcripts of the 8473T allele and promote mRNA decay , whereas the cancer-associated variant 8473C allele interfered with miR-542-3p binding. Colon cancer cells and tissue displayed COX-2 expression levels that were dependent on T8473C allele dosage and allelic-specific expression of COX-2 was observed to be a contributing factor promoting COX-2 overexpression (Moore et al. 2012). These findings provide a novel molecular explanation underlying cancer susceptibility associated with COX-2 T8473C SNP and suggest that other SNPs might influence miRNA activity involved in PG synthesis.
2.3.11 miR-338
A relationship between miR-338 loss and several cancers including gastric, colorectal, and neuroblastoma has been found (Peng et al. 2014; Chen et al. 2013; Sun et al. 2014). MiR-338 plays an important regulatory role in PG synthesis by targeting directly PLA2G4B (Phospholipase A2 Group VI B), a member of the cPLA2 family in human chorioamniotic membrane (Montenegro et al. 2009). The downregulation of miR-388 at term is associated with an induction of PLA2G4B and inhibition of miR-338 leads to an induction of PLA2G4B in decidual cells (Montenegro et al. 2009).
2.3.12 Targeting miRNAs as a Therapeutic Approach to Restore Prostaglandin Homeostasis
Altered prostaglandin homeostasis in various pathologies has driven various therapeutic strategies aimed at targeting the activity of the key enzymes involved in this process. In this context, several pharmacologic inhibitors specifically targeting the activity of COX-2 have been developed (e.g. celecoxib, rofecoxib). However, this therapeutic approach revealed also several off-target and severe side effects, such as an increased risk of myocardial infarction, gastric ulceritis or hepatic toxicity that limit clinical feasibility (Tegeder et al. 2001; Tan et al. 2007; Steffel et al. 2006; Dogne et al. 2006). More recently, it has been suggested that targeting the expression of COX-2, rather than its activity would be a better approach with less side effects (Cerella et al. 2010). An attractive alternative strategy may reside in modulating the expression or activity of pathogenic miRNAs involved in promoting altered prostaglandin homeostasis. Moreover, the ability of some miRNAs to target multiple players in the PG pathway would allow an efficient inhibition of PGs synthesis. Recent findings have shown that chemically modified synthetic nucleotides mimicking or inhibiting endogenous miRNAs can be used for therapeutic purpose (van Rooij et al. 2012; Stenvang et al. 2012; Thorsen et al. 2012; Garzon et al. 2010; Deiters 2010). Antisense modified oligonucleotides (AMOs) (2′-O-methoxyethyl phosphorothioate modified antisense oligonucleotide; 2′-Fluoro modified antisense oligonucleotide), antagomiRs (3′cholesterol-conjugated, 2′-O-Me oligonucleotides having terminal phosphorothioate), and locked nucleic acid (LNA) modifications display increased affinity for their targets and are stable in serum allowing for in vivo delivery (Stenvang et al. 2012). Delivery of synthetic oligonucleotides has shown in vivo feasibility through intravenous or intraperitoneal administration. Miravirsen, a locked nucleic acid–modified DNA phosphorothioate antisense oligonucleotide targeting miR-122 , was the first miRNA-targeted drug to enter clinical trials in 2008 for the treatment of HCV by intravenous injection (Hu et al. 2012). Other studies have demonstrated the ability of new formulated 2′-O-(2-methoxyethyl) modified antisense oligonucleotide (2′-MOE ASO) combined with a permeation enhancer (sodium caprate) for oral administration in humans (Tillman et al. 2008). In addition, special formulations have been made in order to improve the delivery of oligonucleotides such as liposomal or oleic-based nanoparticles formulation (Trajkovski et al. 2011; Wang et al. 2013b).
Several molecules from natural origins and particularly dietary polyphenols have the ability to modulate the expression of miRNAs involved in various pathologies. These compounds, such as curcumin and coffee polyphenols, can be found in many fruits and vegetables and display beneficial effects in various disorders via oral delivery (Prasad et al. 2014; Murase et al. 2011). Some of these molecules can modulate the expression of specific miRNAs involved in PGs biosynthesis. For instance, epigallocatechin gallate (EGCG), from green tea upregulates miR-16 in hepatocellular carcinoma cells (Tsang and Kwok 2010).
2.4 Processing Bodies and Stress Granules
Processing bodies (P-bodies) and stress granules (SG) are RNA granules that play an integral role in mRNA turnover and translational regulation. Although P-bodies were originally identified as cytoplasmic sites of mRNA degradation , evidence indicates that it can also serve as sites of short-term mRNA storage (Eulalio et al. 2007; Garneau et al. 2007). Similarly, SGs serve as interim sites of mRNA storage in cells subjected to severe stress. Both P-bodies and SGs are non-membrane bound granula whose formation is dynamic and reversible. Immunofluorescence analysis demonstrates that, in response to stress, P-bodies and SGs exist in close proximity and partial co-localize. This aspect supports the hypothesis that transcripts traffic between P-bodies and SGs, and that irreversible stresses may result in shuttling of mRNA transcripts from SGs to P-bodies for subsequent decay (Kedersha et al. 2005; Anderson and Kedersha 2008).
Since their discovery, many studies have catalogued the core components that comprise these RNA granules. P-bodies harbor enzymes responsible for mRNA decapping (e.g., DCP1a, DCP2, HEDLS), mRNA deadenylation (CAF-1, CCR4) and degradation (XRN-1), miRNA decay machinery (e.g., AGO2), and components of nonsense-mediated mRNA decay (NMD) (Eulalio et al. 2007; Kulkarni et al. 2010; Zheng et al. 2011) (Fig. 9.4). SGs are comprised of largely stalled translational pre-initiation complexes, small ribosomal subunits (e.g., eIF3, eIF4E, eIF4G) and their core components (Kedersha and Anderson 2007). In addition to these core components, several ARE -binding proteins have been shown to transiently localize to P-bodies and SGs, allowing for delivering of their mRNA target for translational suppression and/or decay (Blanco et al. 2014; Franks and Lykke-Andersen 2007; Gilks et al. 2004). Recently it has been shown that ARE-binding proteins play an important role in the control of P-body formation. Induction of TTP expression by the growth-inhibitory cytokine TGF-β results in enhanced P-body formation, resulting in TTP-driven sequestration of the COX-2 mRNA transcript to P-bodies (Blanco et al. 2014). This ability of TTP to promote P-body formation was also observed in vivo, where analysis of colonic epithelium in TTP knockout mice showed limited numbers of P-bodies per cell as compared to wild-type littermates. Similarly, fibroblasts derived from these mice indicate that absence of TTP expression dramatically impairs P-body formation (Blanco et al. 2014). In line with these findings, loss of TTP expression in epithelial tumors correlates with low cellular number of P-bodies. These findings provide mechanistic insights into how TTP promotes the decay of COX-2 mRNA and controls prostaglandin formation in normal intestinal epithelium.
Related posts:
 Translational and Post-translational Control of Leptin Production by Fat Cells
Translational and Post-translational Control of Leptin Production by Fat Cells
 MicroRNA Regulation of Endocrine Functions in the Ovary
MicroRNA Regulation of Endocrine Functions in the Ovary
 Post-transcriptional and Post-translational Regulation of Steroidogenesis
Post-transcriptional and Post-translational Regulation of Steroidogenesis
 Post-transcriptional Regulation of Insulin and Insulin Like Growth Factors
Post-transcriptional Regulation of Insulin and Insulin Like Growth Factors
 Mechanisms of Post-transcriptional Gene Regulation
Mechanisms of Post-transcriptional Gene Regulation
 Post-transcriptional Regulation of Parathyroid Hormone Gene Expression in Health and Disease
Post-transcriptional Regulation of Parathyroid Hormone Gene Expression in Health and Disease
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


