Fig. 4.1
LHR mRNA levels during hCG-induced downregulation. Northern blot analysis of steady state LHR mRNA levels in total RNA, isolated at the indicated times, from the ovaries of saline-injected (control) (lanes 1–6) or hCG-injected (downregulated) (lanes 7–11) rats. Blots were probed using a labeled cDNA encoding the LHR carboxyl terminus and a portion of the 3′-UTR (nucleotides 1936–2682). RNA was extracted from control and downregulated ovaries at 0, 6, 12, 24, 48, and 72 h after hCG injection. This research was originally published in J Biol Chem (Kash JC, Menon KMJ. J Biol Chem. 1998. 273:10658–64)
3 Accelerated LH Receptor mRNA Degradation as a Regulatory Mechanism
It is now well recognized that the expression of specific, highly regulated mRNAs is regulated, at least in part, at the level of mRNA degradation . There are several examples where the mRNA degradation rate controls the steady state levels of mRNA expression (Port et al. 1992; Bernstein et al. 1992; Sachs 1993; Shyu et al. 1989; Ross 1995; Moghul et al. 1994; Olivas and Parker 2000; Tharun et al. 2000; Pachter et al. 1987; Perez-Ortin et al. 2013; Garneau et al. 2007). In almost all instances where the mRNA expression is regulated by controlling its degradation, the changes in the stability of the mRNA have been shown to result from the binding of specific proteins to specific sequences and/or structures of the mRNA. The specific regions that the mRNA binding proteins interact with may be localized either on the 5′ untranslated region, the coding region or the 3′ untranslated region of the mRNA (Ross 1995). In general, the steady state levels of the mRNA expression are regulated by either increasing or decreasing the degradation rate resulting from the interaction with specific RNA binding proteins .
The possible existence of a binding protein that specifically recognizes LHR mRNA was examined by incubating [32P] labeled LH receptor mRNA with a cytosolic fraction (100 × g supernatant) prepared from the ovary pretreated with hCG to downregulate LH receptor mRNA expression in the presence of ribonuclease inhibitors (Kash and Menon 1998, 1999). After treating the reaction mixture with ribonuclease T1 to degrade the unreacted RNA probe, and the ribonucleoprotein (RNP) complex was separated by electrophoresis on an 8 % native acrylamide gel and subjected to autoradiography. Two RNP complexes were identified, one prominent band corresponding to 50 kDa and a second less intense band of 45 kDa (Fig. 4.2). A threefold increase in the 50 kDa RNP complex was seen in samples derived from LHR downregulated ovaries but no significant change in the intensity of the 45 kDa band was found. Because of this reason, further studies focused on the larger 50 kDa ribonucleoprotein complex. The protein was named LH receptor mRNA binding protein and abbreviated as LRBP (Kash and Menon 1998, 1999).
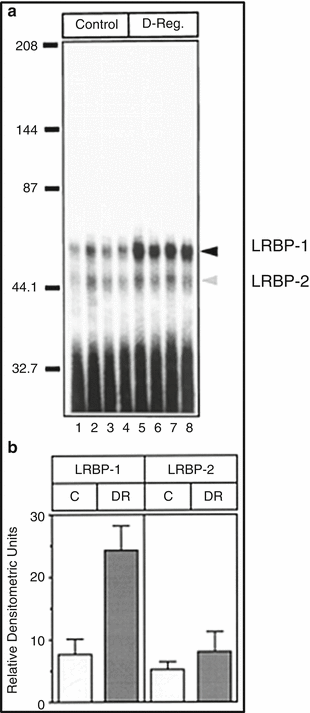
Fig. 4.2
Binding of LHR mRNA to LRBP -1 and LRBP-2 during hCG-induced downregulation. Ovaries were collected from saline-injected (Control) and hCG (12 h) downregulated (D-Reg.) rats. S100s from the control (lanes 1–4) and hCG downregulated (lanes 5–8) ovaries were subjected to RNA gel mobility shift analysis by incubation with radiolabeled (1 × 105 cpm) LHR RNA followed by 8 % native polyacrylamide gel electrophoresis. (a) Autoradiogram of gel shift analysis. Arrows indicate LRBP-1 and LRBP-2. (b) Bar graph representing the mean densitometric scans ± S.D. of the LRBP-1 and LRBP-2 complexes shown in (a) (Modified from Kash, J.C. and Menon, K.M.J., 1998, Fig. 6 with permission from the Journal of Biological Chemistry)
The binding of LHR mRNA to LRBP showed that the recognition of the mRNA resided in the amino terminal portion of the LH receptor corresponding to nucleotides 102–282. The RNP complex formation was always higher in the ovarian tissue derived from LH receptor downregulated ovaries with no change in the non-target tissues (Kash and Menon 1998). Further truncation of this region showed that the contact site resided between nucleotides 188–228. RNA hydroxy-radical foot-printing revealed a bipartite polypyrimidine rich sequence, (UCUCX7-UCUCCCU) corresponding to nucleotides 203–220 of LHR mRNA that specifically interacted with LRBP (Fig. 4.3). Mutation of the C residues within the bipartite sequence revealed that all C residues participated in binding to LRBP (Kash and Menon 1999). The contact site was further confirmed by performing RNA electrophoretic mobility shift assays. The binding was specific, since the RNP complex formation using [32P] labeled LH receptor mRNA probe (nucleotides 203–220) was not displaced by probes corresponding to other regions of LH receptor mRNA. The interaction of LHR mRNA with LRBP exhibited high affinity with equilibrium dissociation constant (Kd) in the range of 4 × 10−9 M.
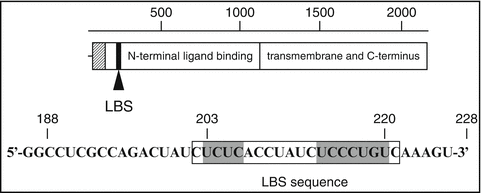
Fig. 4.3
Diagram of the LRBP RNA binding site. LH/hCG receptor mRNA open reading frame showing the position of the LRBP binding site (LBS). The sequence of the LHR:188–228 is also shown (Modified from Kash, J.C. and Menon, K.M.J., 1999, Fig. 8 with permission from Biochemistry)
After establishing that the ovarian LRBP recognizes LH receptor mRNA with high affinity and specificity, the changes in LRBP activity in relation to tissue levels of LH receptor mRNA were examined. It has been established that LHR mRNA expression increases during maturation of the ovarian follicles in response to FSH. The levels then fall immediately after the preovulatory LH surge and the levels rise again with the growth and development of the corpus luteum. The relationship between LHR mRNA expression and LRBP activity was examined in ovarian tissues after treating immature rats with pregnant mare serum gonadotropin to induce follicle growth and to increase LHR mRNA expression levels. At this time point, the LRBP activity in the S100 fractions prepared from the ovaries showed low levels of activity. Thus a negative correlation between tissue levels of LHR mRNA expression and LRBP activity was observed. However, when LHR expression was suppressed during ligand-induced downregulation by treatment with hCG, the LRBP activity was increased several fold in a time dependent manner. Thus, when the LH receptor mRNA expression level was high, the RNA binding protein activity was at the lowest level. Conversely, the LH receptor mRNA binding activity was high when the mRNA levels began to fall. This inverse relationship suggested that LRBP is an endogenous regulator of LH receptor mRNA expression.
4 Signaling Pathways Involved in Ligand-Mediated Downregulation of LH Receptor mRNA
Since the immediate response of the ovarian tissue to hCG treatment is to increase the production of cyclic AMP (Menon and Gunaga 1974; Marsh et al. 1966), the involvement of cyclic AMP in LHR mRNA downregulation was presumed. In fact, this assumption turned out to be true by the demonstration that chronic elevation in cyclic AMP production in ovaries of superovulated rats with multiple injections of rolipram, an inhibitor of type 2 phosphodiesterase (Peegel et al. 2005), showed downregulation of LH receptor mRNA similar to that produced by the treatment with hCG. The ovaries were then processed for REMSA in order to determine whether there was increased LRBP activity when LH receptor mRNA was downregulated. A substantial increase in the RNA binding activity was seen in ovarian S100 fractions at time intervals when the LH receptor mRNA expression was downregulated. These findings suggested that the downregulation of LH receptor mRNA in response to hCG treatment was caused by an increase in cyclic AMP production. In addition, we were able to demonstrate that cyclic AMP-mediated activation of PKA and ERK1/2 signaling pathways is involved in ligand-mediated LH receptor downregulation in human granulosa cells (Menon et al. 2011). The participation of PKA and ERK1/2 signaling pathways was demonstrated with the use of H89 and U0126, respectively. Furthermore, transfecting cultured human granulosa cells with ERK1/2-specific small interfering RNA abrogated LH receptor downregulation in response to hCG treatment thereby conclusively demonstrating the participation of ERK1/2 signaling pathways in inducing downregulation (Menon et al. 2011).
5 In Vitro mRNA Degradation by the LH Receptor mRNA Binding Protein
After establishing a relationship between the expression of LH receptor mRNA and the RNA binding protein under in vivo conditions, the direct role of the RNA binding protein in LH receptor mRNA degradation was demonstrated by employing a cell-free mRNA decay system developed by Ross and colleagues (Ross 1993) under in vitro conditions. The assay essentially determines the degradation rate of a specific exogenous RNA in the presence and absence of the RNA binding protein under controlled conditions. Although all mRNAs are prone to degradation, the rate of degradation varies depending on the cellular environment. Some mRNAs are more labile than others, a property conferred by the presence of appropriate trans-acting factors that interact with specific structures or sequences present in the mRNA. Initially, we showed that the rate of decay of LH receptor mRNA was very rapid in ribosomes isolated from the ovaries of rats treated with hCG, compared to the degradation of LH receptor mRNA by ribosomes isolated from ovaries of saline treated control animals (Nair et al. 2002), but the rate of decay of exogenously added ovarian RNA by ribosomes isolated from saline treated rats was accelerated by the addition of a partially purified ovarian LH receptor mRNA binding protein (Nair et al. 2002). The rate of LH receptor mRNA decay was not affected by unrelated proteins added to the reaction mixture. These experiments demonstrated that the LHR mRNA binding protein plays a role in LH receptor mRNA degradation.
6 Characterization of LH Receptor mRNA Binding Protein
Purification of RNA binding proteins from tissue samples presents many challenges including the relatively time consuming assays to follow the RNA binding activity using RNA electrophoretic gel mobility shift assay and its low abundance in the cell. Covalently linked RNA binding sequence to sepharose followed by column chromatography showed that the binding protein was retained on the columns, but the attempts to elute the binding protein retained on the matrix were not successful. Therefore, conventional techniques were used to purify the LH receptor mRNA binding protein from the 100 × g supernatant fraction of the ovarian homogenates. The ovaries were initially downregulated by treatment with the ligand in order to increase the yield of the binding protein. The supernatants were subjected to chromatography on a strong cation exchange resin (Macro-Prep high S support) and eluted with 150 mM KCl. The eluates were concentrated and subjected to SDS-PAGE to separate the proteins. The [32P] LHR mRNA binding activity associated with the protein band on the gel was identified by an overlay assay (Northwestern blot) using [32P] labeled LH receptor mRNA fragment (203–220) as the probe. After extensive standardization of the assay, the protein band corresponding to the band that showed the RNA binding activity was cut, eluted and renatured. The eluted protein was electrophoresed again to determine the purity of the preparation. The electrophoretically homogeneous protein band was then subjected to amino terminal analysis as well as MS-MALDI analysis to establish its identity. Both analyses revealed the purified protein to be mevalonate kinase (Nair and Menon 2004). The gene encoding rat mevalonate kinase was then cloned and overexpressed in 293 T cells (Nair and Menon 2004). The recombinant protein exhibited a concentration dependent increase in binding LHR mRNA probe (Nair and Menon 2004). The binding exhibited all the characteristics of the expected LHR mRNA binding protein with respect to specificity for binding to the previously identified contact site (nucleotides 203–220), competition by unlabeled LHR mRNA fragment (203–220), dependence on C residues in the ligand binding site and immunoreactivity of the recombinant protein similar to that seen for the electrophoretically purified rat mevalonate kinase by Western blot analysis. Furthermore, since mevalonate kinase is known to have two binding sites, one for ATP and a second site for mevalonate, the involvement of these sites in LH receptor mRNA binding activity was then determined. It was seen that the binding of the [32P] labeled LH receptor mRNA binding sequence to mevalonate kinase was inhibited by ATP and mevalonate. The inhibitory effect was even more pronounced in the presence of both ATP and mevalonate. These results clearly confirm the identity of the RNA binding protein as mevalonate kinase. Mevalonate kinase is a metabolic enzyme involved in cholesterol biosynthesis which catalyzes the conversion of mevalonate to phosphomevalonate, the precursor of cholesterol and many natural products. In fact, we have shown that mevalonate kinase expression is regulated by LH in the corpus luteum both at the mRNA level as well as at the protein level (Wang and Menon 2005). During LH/hCG-induced downregulation, mevalonate kinase expression was induced in the corpus luteum. The induction of expression of other SRE containing genes such as HMG CoA reductase, farnesyl pyrophosphate synthase and LDL receptor were also seen during this phase (Wang and Menon 2005). These results suggest that mevalonate kinase expression plays a regulatory role in LH receptor mRNA expression in the corpus luteum.
Depletion of mevalonate kinase expression by 25 hydroxycholesterol resulted in the abrogation of LH receptor mRNA downregulation in cultured human granulosa cells (Menon et al. 2011; Wang and Menon 2005). The inhibitory effect of mevalonate kinase on LH receptor expression was documented by Ikeda et al. 2008 who showed that overexpression of mevalonate kinase can abrogate FSH-induced increase in LH receptor mRNA expression in rat ovarian follicles.
The notion that mevalonate kinase , a metabolic enzyme, acts as an mRNA binding protein is consistent with a similar role of several other metabolic enzymes that have been recently described (Menon et al. 2005; Hentze 1994). The iron response element binding protein, which plays a role in iron homeostasis in cells, is an enzyme in the citric acid cycle. Similarly, thymidylate synthase, dihydrofolate reductase, glyceraldehyde-3-phosphate dehydrogenase, glutamate dehydrogenase and lactate dehydrogenase all have been shown to serve as RNA binding proteins that regulate different aspects of RNA metabolism. In the case of mevalonate kinase, structural studies have shown that it belongs to a family of ATP binding proteins containing a conserved glycine motif. Members of this group include galactokinase, homoserine kinase, mevalonate kinase and phosphomevalonate kinase. These proteins share a common fold, a left handed β-α-β loop known as the ribosomal protein S5 domain 2 like fold. This fold is similar to that found in proteins that interact with nucleic acids including elongation factor G and ribonuclease P (Zhou et al. 2000). Thus from a structural point, it is conceivable that mevalonate kinase can serve as an LH receptor mRNA binding protein to regulate cellular expression of LH receptor under select conditions.
7 Mechanism of LH Receptor mRNA Decay by LRBP
The endogenous association of LRBP with LH receptor mRNA was seen during downregulation supporting the view that LRBP is likely responsible for rapid degradation of LHR (Nair et al. 2002). The evidence comes from studies using ovarian extracts from the downregulated ovaries. The RNP complexes isolated from these ovaries were immunoprecipitated with anti LRBP antibodies and RNA extracted from the immunoprecipitates. RNAs were reverse transcribed and then subjected to PCR amplification using specific probes for LH receptor. Significant amplification of LHR was seen in the downregulated ovarian extracts suggesting that the association of LRBP with LHR mRNA occurs during downregulation (Nair et al. 2002). Since the coding region of LHR mRNA is recognized by LRBP, it is quite likely that LRBP might act as a translational suppressor of LHR mRNA. This was indeed the case. We developed a cell free protein synthesizing system in which FLAG tagged LHR mRNAs were efficiently translated using an in vitro rabbit reticulocyte lysate system to yield 63 kDa protein species corresponding to non-glycosylated LHR. Addition of purified LRBP blocked the translation of LH receptor mRNA while addition of a region of corresponding to LRBP binding sequence (LBS) in which all the cytidine residues were mutated to uridine residues were not affected (Fig. 4.4). The wild type LBS effectively inhibited the translation of LH receptor mRNA. This clearly showed that LRBP acts as a translational suppressor of LH receptor mRNA and the loss of LH receptor mRNA during downregulation suggested that untranslatable LH receptor mRNA might be targeted for degradation. The translation of eukaryotic mRNAs is a complex process. In the cytoplasm, the fully processed mRNA can exist either in a polysome-associated, translationally active form or in an inactive messenger ribonucleoprotein (RNP) complex (Xu et al. 2000; Xu and Grabowski 1999; Mathews et al. 2000; Hershey 1991). The exchange between these two compartments modulates the fraction of mRNA available for translation (Kozak 1989). A clear link between translation and decay are intimately connected since the factors and processes required for mRNA translation and decay are closely connected (Ross 1995; Mathews et al. 2000; Jacobson and Peltz 1996) and translational arrest, in general, alters the degradation rate of most eukaryotic mRNAs (Ross 1995; Mathews et al. 2000).
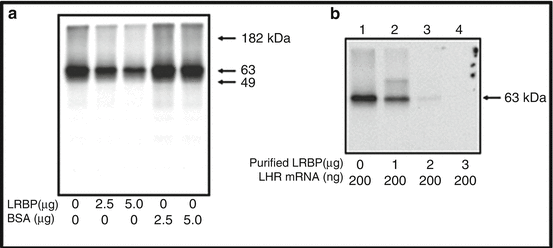
Fig. 4.4
Translational suppression of LHR mRNA by LRBP in vitro. (a) FLAG-tagged rat LHR mRNA (200 ng) was in vitro translated using a Flexi rabbit reticulocyte lysate system in the presence of [35S] methionine and partially purified rat LRBP from the ovary or fatty acid-free bovine serum albumin (BSA) at different concentrations (2.5 and 5.0 μg). The translated LHR protein was immunoprecipitated with anti-FLAG antibody, separated on 10 % SDS-PAGE, and processed for autoradiography. (b) In vitro translation of FLAG-tagged rat LHR mRNA using [35S]methionine in the presence of increasing concentrations (1, 2, and 3 μg) of purified rat LRBP, followed by immunoprecipitation, SDS-PAGE and autoradiogram. This research was originally published in J Biol Chem. (Nair AK et al. J Biol Chem. 2005, 280, 42809–42816)
Since LRBP is a cytosolic protein, we further examined its translocation from cytoplasm to ribosomes during downregulation to cause translational suppression . We were able to show that Western blot analysis of ribosome rich fractions from FPLC-assisted gel filtration of post-mitochondrial fractions showed the presence of LRBP in translating ribosomes from Downregulated ovaries , but not from the control ovaries. Increased association of LH receptor mRNA with LRBP was seen in the polysomes isolated from the downregulated ovaries when compared to the control group. The increased association of LH receptor mRNA with LRBP in polysomes from downregulated ovaries was associated with decreased translational effectiveness of these polysomes again showing the translational suppression by LRBP (Menon et al. 2009). Thus, during downregulation, an increase in the translocation of LRBP to ribosome and its increased association with LHR mRNA leads to its increased degradation.
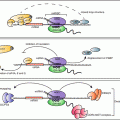
Increased translocation of LRBP to polysome and increased association in translating ribosomes during downregulation suggested that these processes precede a decrease in LH receptor mRNA levels. The effect of these early events during downregulation showed that the association of LRBP with ribosomes and translational suppression led to its rapid degradation by facilitating decapping . Immunoprecipitation of the complex with 5′ cap structure antibody followed by real time PCR analysis using specific LH receptor mRNA probes revealed progressive loss of capped LH receptor mRNA during downregulation (Menon et al. 2013a). RNA immunoprecipitation confirmed the dissociation of eukaryotic initiation factor 4E from the cap structure which is required for decapping. Furthermore, RNA immunoprecipitation with antibody against the P body marker protein, DCP1A revealed that LH receptor mRNA was associated with the P bodies, the cytoplasmic foci that contain RNA degradative enzymes and decapping complex (Menon et al. 2013a). Interestingly, immunohistochemical studies using antibodies against LRBP and DCP1A followed by confocal analysis showed co-localization of LRBP with DCP1A during LHR downregulation. The association of LRBP and LH receptor mRNA in the P bodies during downregulation was also confirmed by examining the association of a second P body marker rck/p54. These results are supportive of the notion that during downregulation, LRBP forms an untranslatable ribonucleoprotein complex which is then translocated to P bodies leading to decapping and degradation by exonucleases (Fig. 4.5).
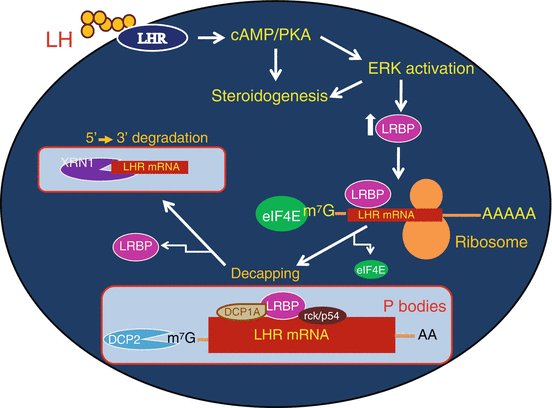
Fig. 4.5
Proposed mechanism of LRBP mediated degradation of LHR mRNA in the P bodies. LH signaling through its receptor activates cAMP/PKA/ERK pathways, leading to increased levels of LRBP. LRBP binds to the coding region of LHR mRNA in the ribosomes and an untranslatable ribonucleoprotein complex is formed. LRBP also associates with the P body components such as DCP1A and rck/p54 resulting in the P body localization of this mRNP complex. This is followed by the dissociation of eIF4E from the cap structure and the decapping of LHR mRNA in the P bodies, initiated by DCP2 enzyme. Final step is the 5′-3′ exonuclease (XRN1) mediated degradation of LHR mRNA
8 Interaction of LRBP with Other Trans Factors during Downregulation
Since LRBP is devoid of any nuclease activity capable of degrading LH receptor mRNA, the possibility of other proteins that selectively interact with LRBP and if such proteins exist, their functional roles in directing LH receptor mRNA to decay pathways was examined. We used a yeast two hybrid system to screen cDNA library which was constructed from LHR downregulated ovaries (Wang et al. 2010). Using this strategy we identified five potential candidate proteins that interacted with LRBP during downregulation. Among these, UBCE2i which is now recognized as the sumoylating enzyme has been implicated in nuclear-cytoplasmic transport in addition to other cellular functions. SUMOplotT revealed that LRBP has two potential sumoylation sites at K 345 and K 256. Overexpressed LRBP in 293 T cells was assayed for sumoylation (Wang et al. 2010) and revealed that both lysine 256 and 345 were sumoylated whereas mutation of the lysine residues to arginine blocked sumoylation. Although the actual functional significance of sumoylation of LRBP is not understood, it is likely that sumoylation might play a role in the translocation of LRBP-bound LH receptor mRNA to P bodies for degradation.
Related posts:
 Translational and Post-translational Control of Leptin Production by Fat Cells
Translational and Post-translational Control of Leptin Production by Fat Cells
 MicroRNA Regulation of Endocrine Functions in the Ovary
MicroRNA Regulation of Endocrine Functions in the Ovary
 Post-transcriptional and Post-translational Regulation of Steroidogenesis
Post-transcriptional and Post-translational Regulation of Steroidogenesis
 Post-transcriptional Regulation of Prostaglandin Biosynthesis
Post-transcriptional Regulation of Prostaglandin Biosynthesis
 Post-transcriptional Regulation of Insulin and Insulin Like Growth Factors
Post-transcriptional Regulation of Insulin and Insulin Like Growth Factors
 Mechanisms of Post-transcriptional Gene Regulation
Mechanisms of Post-transcriptional Gene Regulation
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


