Fig. 2.1
Schematic representation of the human insulin (a) and mouse insulin 2 (b) genes. The both genes consist of three exons (boxes), and two introns (lines). The coding region shown as gray boxes starts in exon 2 and the arrows indicate the translation start sites. Intron 1 in both the human insulin (a) and mouse insulin 2 (b) genes is flanked by the canonical 5′-splice site sequence AG/GT and the 3′-splice site AG. The arrowheads indicate the 5′-alternative splice site within intron 1 in the human insulin gene (a) and 3′-alternative splice site within exon 2 in the mouse insulin 2 gene (b). The 5′ UTR shown as white boxes includes exon 1 and part of exon 2. In humans, usage of the 5′-alternative splice site results in retention of the first 26 bp of intron 1 (a), while, in mouse, usage of the 3′-alternative splice site results in loss of the first 12 bp of exon 2 without altering the coding sequence (b) (Modified from Shalev et al. 2002 and Panda et al. 2010)
2.2 Post-transcriptional Regulation of INS mRNA by miRNAs
The discovery of miRNAs has added a novel regulatory layer to post-transcriptional control of INS mRNA in pancreatic β-cells. The major miRNAs reported to play a direct role in this process in pancreatic β-cells are shown in Table 2.1 and have been discussed below.
Table 2.1
Post-transcriptional regulators of insulin and insulin-like growth factors
Target mRNA | Regulators | INS/IGF levels | Function | Reference |
INS | HuD | Down | Translational reppression of insulin mRNA | Lee and Kim (2012) |
PTB | Up | Glucose-induced stabilization of insulin mRNA | Tillmar L (2002) | |
PDI and PABP | Up | Translational activation of insulin mRNA | Kulkarni SD (2011) | |
miR-196b | Up | Translational activation of insulin mRNA | Panda AC (2014) | |
miR-25 and -92 | Down | Destabilization of insulin mRNA | Karolina DS (2013) | |
IGF-II | Zcchc11 | Up | up-regulation of IGF-I by uridylation of IGF-I targeting miRNAs | Joens MR (2012) |
Nocturnin | Down | destabilizing of long form of IGF-I mRNA by deadenylation | Kawai M (2010) | |
SF2/ASF | Exon 5 inclusion during myogenic differentiation, suclse hypertrophy, and myopathy | Smith PJ (2002) | ||
miR-1 | Down | Translational inhibition of IGF-I in glucose-induced apoptosis | Yu X (2008) | |
miR-1 | Down | Translational inhibition of IGF-I in cartilage cell proliferation | Hu X (2013) | |
miR-1 | Down | Translational inhibition of IGF-I in muscle cell and inhibition of IGF signaling | Elia L (2009) | |
miR-1/206 | Down | Promotion of apoptosis after myocardiac infraction | Shan ZX (2009) | |
miR-29 | Down | promotion of myoblastic transition of HSC | Kwiecinski M (2012) | |
miR-29 | Down | inhibition of IGF-I signaling in aged brain | Fenn AM (2013) | |
IGF-II | IMPs | Down | translational reppression of IGF-II L3 mRNA during embryonic development | Nielsen J (1999) |
IMP3 | Up | translational activation of IGF-II L3 mRNA in K562m glioblastoma | Liao B (2005, 2011) | |
IMP2 | Up | IRES translation activation of IGF-II L3 mRNA during embryogenesis | Dai N (2011) | |
Lin-28 | Up | Translational activation of IGF-II and promotion of myogenesis | Polesskaya A (2007) | |
miR-125b | Down | Translational inhibition of IGF-II and inhibition of muscle differentiation | Ge Y (2011) | |
miR-100 | Down | Translational inhibition of IGF-II and inhibition of cell ;proliferation and survival signaing in breast cancer | Gebeshuber CA (2013) |
2.2.1 miR-25 and 92a
In pancreatic β-cells, miRNAs regulate glucose-stimulated insulin secretion and β-cell survival as well as insulin biosynthesis (Kim and Lee 2012). Recently, the Jeyaseelan group reported two miRNAs (miR-25 and miR-92a) as direct regulators of insulin biosynthesis at the mRNA level (Setyowati Karolina et al. 2013). The 3′ UTR of INS mRNA contains binding sites for miR-25 and 92a that are conserved in the rat and mouse and are partially conserved in humans. Overexpression of miR-25 or miR-92a not only reduced INS mRNA but also its biosynthesis; conversely, introduction of anti-miR-25 or miR-92a increases insulin biosynthesis. Interestingly, significant up-regulation of miR-25 and miR92a is observed in the pancreas of diabetic rats, in which INS mRNA levels are decreased.
2.2.2 miR-196b
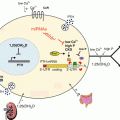
Although miRNAs typically target the 3′ UTR of the mRNA for functional inhibition(Pillai et al. 2007; Yekta et al. 2004), they can also target the 5′ UTR and coding regions of mRNA as well as increase translation of target mRNAs through the 3′ and 5′ UTR (Zhou et al. 2009; Orom et al. 2008; Vasudevan et al. 2007). Most recently, the Seshadri group has reported that miR-196b can directly increase INS mRNA translation by targeting the 5′ UTR (Panda et al. 2014). Mouse miR-196b specifically targets the 5′ UTR of INS mRNA and the binding site of miR-196b overlaps with that of HuD, a 5′ UTR-associated RBP that represses INS mRNA translation. Through this binding, miR-196b increases INS mRNA translation and this is likely Ago2-dependent process, because miR-196b-mediated activation of insulin expression is abolished when Ago2 levels are depleted (Panda et al. 2014). Interestingly, suppression of miR-196b expression causes increased association of HuD with the 5′ UTR of INS mRNA and, conversely, this association is modestly decreased by overexpression of miR-196b, probably due to altered stem-loop structure of the 5′ UTR. HuD and miR-196b might compete for binding to the same site in the 5′ UTR of INS mRNA, providing an example of coordinated regulation of translation by miRNA and RBP (Fig. 2.2).
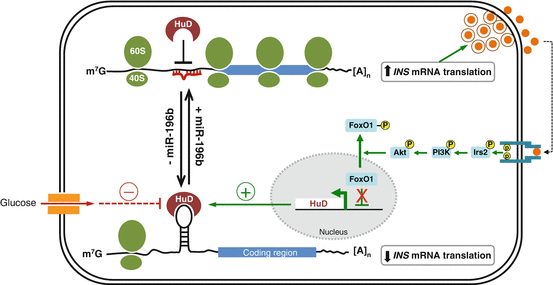
Fig. 2.2
A proposed model of HuD and miR-196b action on insulin biosynthesis
2.3 Post-transcriptional Regulation of INS mRNA by RBPs
Despite the short UTRs of INS mRNA, post-transcriptional regulation of INS mRNA is mediated through the cooperative action of a stem-loop in the 5′ UTR and the conserved UUGAA sequence in the 3′ UTR (Wicksteed et al. 2001). However, the identities of the specific factor(s) that associate with these elements had remained unclear until recently. Recently, several RBPs in pancreatic β-cells have been identified as pivotal post-transcriptional regulators of INS mRNA due to their effects on its stability and translation.
2.3.1 HuD
The Hu/ELAV (human antigen/embryonic lethal abnormal vision-like) family includes the ubiquitously expressed HuR (HuA) and the primarily neuronal HuB, HuC, and HuD. HuD/ELAVL4 is reported to specifically and directly affect post-transcriptional regulation of INS mRNA (Kim and Lee 2012). Like HuB and HuC, HuD expression was believed to be restricted primarily to neurons (Hinman and Lou 2008). However, recently it has been shown to be expressed in insulin -producing pancreatic β-cells (Lee et al. 2012; Poy et al. 2009), in which its levels are controlled by the insulin signaling pathway, sequentially implicating Irs2, PI3K, Akt, and FoxO1 (Fig. 2.2). Like other Hu/ELAV family proteins, HuD controls stability and translation of target mRNAs by binding to 5′ or 3′ UTR bearing AU- and U-rich sequences through three highly conserved RNA recognition motifs (RRMs) (Hinman and Lou 2008; Pascale et al. 2008).
In pancreatic β-cells, HuD, but not HuR and other RBPs, including HuR, TIAR [T-cell-restricted intracellular antigen-1 (TIA-1)-related protein], heterogeneous nuclear ribonucleoprotein (hnRNP) K, and nuclear factor (NF) 90, as well as hnRNP C, Ago, and the fragile X syndrome protein (FMRP ), associates with a 22-nucleotide segment in the 5′ UTR , but not with the coding region (CR) or 3′ UTR of mouse INS2 mRNA (Lee et al. 2012). Binding of HuD to the INS2 5′ UTR represses insulin production by reducing the INS2 mRNA translation, and not by altering INS2 mRNA stability . This binding is repressed by glucose treatment, which induces a rapid and robust release of INS2 mRNA from the HuD complex within 30 min of glucose stimulation thereby enables translation of INS2 mRNA (Lee et al. 2012) (Fig. 2.2). Consistently, insulin levels are increased in pancreatic β-cells of HuD-null mice, and conversely, decreased in HuD-transgenic mice. This modulation of insulin by HuD in vivo likely contributes the homeostatic regulation of plasma glucose and insulin concentrations. In support of this is the finding that HuD-transgenic mice display impaired glucose clearance from the blood due to lower plasma insulin levels, thus defining HuD as a pivotal post-transcriptional regulator of insulin.
2.3.2 Polypyrimidine Tract Binding Protein
The polypyrimidine tract-binding protein (PTB ), also known as hnRNP I, binds to pyrimidine-rich sequences of single stranded target mRNAs through four RRMs and plays an important role in several cellular processes such as alternative splicing (Garcia-Blanco et al. 1989; Valcarcel and Gebauer 1997; Wagner and Garcia-Blanco 2001), polyadenylation of pre-mRNAs (Lou et al. 1999; Moreira et al. 1998), cytoplasmic RNA localization (Cote et al. 1999), mRNA stability (Tillmar et al. 2002; Knoch et al. 2004) and translation initiation (Hellen et al. 1993). Glucose-induced stabilization of INS mRNA is a key event in the control of insulin biosynthesis and PTB plays an essential role in this process. In pancreatic β-cells, glucose induces the binding of PTB to the polypyrimidine-rich sequence located in the 3′ UTR of INS mRNA to stabilize INS mRNA, resulting in a glucose-dependent increase in INS mRNA (Tillmar et al. 2002; Tillmar and Welsh 2002). Glucose stimulation of β-cells enhances the cytoplasmic function of PTB by promoting its accumulation in the cytoplasm, upon which PTB binds and stabilizes INS mRNAs (Knoch et al. 2006). The cytoplasmic accumulation of PTB is regulated by cAMP and protein kinase A (PKA)-dependent phosphorylation (Xie et al. 2003; Knoch et al. 2006). Activation of PKA by elevation of cAMP levels causes the direct phosphorylation and nucleo-cytoplasmic transport of PTB (Fig. 2.3). Consistently, glucagon-like peptide 1 (GLP-1), which activates PKA and potentiates glucose-stimulated insulin gene expression and secretion by increasing cAMP levels in β-cells, promotes the phosphorylation of PTB, and conceivably, its cytoplasmic accumulation, which in turn enhances the levels of mRNAs containing PTB binding sites in their 3′ UTR (Knoch et al. 2006). However, GLP-1does not stabilize INS mRNA significantly in either rat insulinoma INS-1 cells or freshly isolated islets (Knoch et al. 2006).
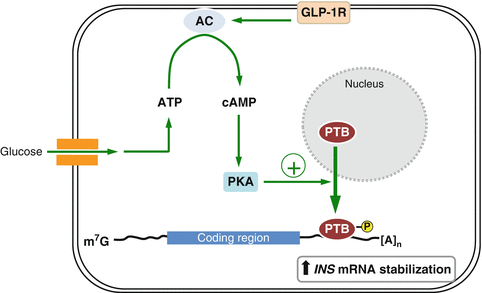
Fig. 2.3
A proposed model of PTB action on insulin biosynthesis
2.3.3 PDI and PABP
Poly(A)-binding protein (PABP ) is a multifunctional RNA binding protein with an N-terminal RNA binding domain composed of four RRMs, and a C-terminal helical domain (Adam et al. 1986), which is important for its interaction with other proteins and for cooperative binding to poly(A) tails (Melo et al. 2003). Through poly(A) tail binding, PABP modulates the stability and translation of target mRNAs (Sonenberg and Dever 2003). Moreover, PABP can also bind to the A-rich sequence in the 5′ UTR to regulate key steps in mRNA translation (de Melo Neto et al. 1995; Oberer et al. 2005). In pancreatic β-cells, PABP binds specifically to the 5′ UTR of INS mRNA and increases the rate of ribosome recycling through its interaction with other translation factors; this leads to higher insulin translation during glucose stimulation (Kulkarni et al. 2011). The binding of PABP to the 5′ UTR of INS mRNA during glucose stimulation is regulated by protein disulfide isomerase (PDI), which was the first identified ER-resident catalyst of native disulfide bond formation (Anfinsen 1973). Glucose stimulation of β-cells induces PDI activation by promoting its phosphorylation. Activated PDI interacts with PABP and catalyzes the reduction of the PABP disulfide bond resulting in specific binding of PABP to the 5′ UTR of INS mRNA and increased insulin translation (Kulkarni et al. 2011).
3 Post-transcriptional Regulation of IGF
Insulin -like growth factor (IGF) is a protein ligand with high sequence similarity to insulin and functions as a key regulator of growth, survival, and differentiation in most cell types (Humbel 1990). There are various mechanisms by which IGF is itself induced and activated, and discrete pathways operate during proliferation, apoptosis, differentiation, and metabolism (King et al. 1982; Panagakos 1993; Zheng et al. 2000). Production of IGF isoforms is tightly regulated at transcriptional, post-transcriptional, and translational levels (Chew et al. 1995; Adamo et al. 1991; Lowe et al. 1989; Hepler et al. 1990; Zhang et al. 1998; Foyt et al. 1991). Dysregulation of IGF levels is responsible for several pathological conditions such as diabetes, cancer, and aging (Cohen and LeRoith 2012). In this section, we review the post-transcriptional regulators of IGFs and their impact on IGF signaling.
3.1 Alternative Splicing of IGF mRNA
Although various splice variants of IGF-I and IGF-II have been characterized (Goldspink and Yang 2004; Velloso and Harridge 2010; Weller et al. 1993; Yang et al. 1995), the factors that govern the spicing factors are largely unknown. The serine-arginine protein splicing factor-1/alternative splicing factor (SF2/ASF) binds to a purine-rich sequence in exon 5 of IGF-I mRNA to promote its inclusion in the mature transcript (Smith et al. 2002). Exon 5 inclusion is responsible for myogenic differentiation, muscle hypertrophy, and myopathy (Barton et al. 2002; Matheny and Nindl 2011). The 5′ UTR composition of IGF-II mRNA varies throughout development due to alternative splicing (Ohlsson et al. 1994; Ekstrom et al. 1995; Monk et al. 2006), although the mechanisms and regulators required for this currently remain uncharacterized.
3.2 Post-transcriptional Regulation of IGF mRNA by miRNAs
3.2.1 Regulation of IGF-I mRNA by miR-1, miR-206, miR-29, and miR-320
IGF-I shows high sequence similarity with insulin and regulates various cellular processes such as cell growth, proliferation, survival, and insulin sensitivity in liver, muscle, and kidney (Lee and Gorospe 2010). Several miRNAs are known to modulate IGF-I expression in different model systems. Among them, miR-1 was shown to target the 3′ UTR of IGF-I mRNA and down-regulate its expression; in cardiomyocytes, miR-1 levels increased during glucose-induced apoptosis and relieved the anti-apoptotic actions of IGF-I (Yu et al. 2008). miR-1 also down-regulated IGF-1 expression in cardiac and skeletal muscle (Elia et al. 2009) and inhibited cell proliferation by directly targeting IGF-I in Chinese sika deer-derived cartilage cells (Hu et al. 2013). Furthermore, miR-1 and miR-206 levels were up-regulated after myocardial infraction, which resulted in a decrease in IGF-I (Shan et al. 2009). Down-regulation of IGF by miR-320 was also reported in myocardial microvascular endothelial cells isolated from type 2 diabetic Goto-Kakizaki rats (Wang et al. 2009). Recently, miR-29a and miR-29b were shown to increase during the myofibroblastic transition of hepatic stellate cells or in the aged brain, resulting in decreased IGF-1 levels (Kwiecinski et al. 2012; Fenn et al. 2013).
3.2.2 Regulation of IGF-II mRNA by miR-100 and miR-125b
IGF-II functions as an embryonic regulator of myogenesis by initiating myogenic differentiation; it is also essential for metastasis of breast cancers (McCann et al. 1996; Pravtcheva and Wise 1998; Florini et al. 1991). IGF-II expression is modulated by miR-125b and miR-100; miR-125b is involved in the down-regulation of IGF-II via direct targeting of 3′ UTR of IGF-II mRNA, and its expression decreases during myoblast differentiation (Ge et al. 2011). miR-125b negatively regulates myoblast differentiation by reducing the IGF-II level. miR-100 also down-regulates IGF-II expression, which leads to the inhibition of cell proliferation and reduces survival signaling in breast cancer cells; consistent with this, miR-100 expression has been found to decrease in human breast cancers (Gebeshuber and Martinez 2013).
3.3 Post-transcriptional Regulation of IGF mRNA by RBPs
3.3.1 Zcchc11
Zcchc11 (zinc finger CCHC domain-containing protein 11, terminal uridyltransferase 4, TUTase4) is a uridyltransferase that can suppress miRNA biogenesis by mediating terminal uridylation of miRNAs , including pre-let-7 and miR-26a (Hagan et al. 2009; Heo et al. 2009; Jones et al. 2009). Zcchc11 down-regulates let-7 in embryonic stem cells by uridylation of pre-let-7 via interaction with Lin28, which is essential for maintaining pluripontency (Hagan et al. 2009). Zcchc11 also catalyzes the 3′ UTR uridylation of miR-26a, thereby promoting expression of cytokines including interlukin-6 (IL-6) (Jones et al. 2009). In Zcchc11-deficient mice, the length and frequency of terminal uridines for diverse mature miRNAs, including miR-126b and miR-379 that target IGF-1 mRNA, decreases (Jones et al. 2012). Uridylation of these miRNAs prevents them from suppressing IGF-I, thereby increasing its expression. Zcchc11 overexpression up-regulates IGF-I level by stabilizing IGF-I mRNA. This reduction in IGF-I level contributes to the increased mortality and reduced growth of Zcchc11-deficient mice because IGF-I is essential for post-natal growth and survival (Jones et al. 2012) (Fig. 2.4).
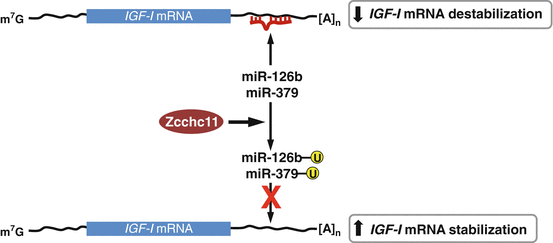
Fig. 2.4
A proposed model of Zcchc11 action on IGF-I biosynthesis
3.3.2 Nocturnin
Nocturnin (Noc, Ccrn4l) is a peripheral circadian-regulated gene product found in the liver, kidney, and testis where it functions as an mRNA deadenylase mediating mRNA degradation (Wang et al. 2001; Baggs and Green 2003; Douris and Green 2008; Kawai et al. 2010). Noc mediates post-transcriptional control of genes necessary for metabolic function including nutrient absorption, insulin sensitivity, lipid metabolism, adipogenesis, inflammation, and osteogenesis (Stubblefield et al. 2012; Green et al. 2007). The protein binds to the long-form 3′ UTR of IGF-I mRNA, which contains potential regulatory motifs involved in IGF-I mRNA degradation (Kawai et al. 2010). Noc overexpression reduces both mRNA and protein levels of IGF-I indicating that Nocturnin enhances IGF-I mRNA degradation, thereby reducing both its mRNA and protein levels. Interestingly, Noc interacts with IGF-I mRNA in a strain- and tissue-specific manner in mice (Kawai et al. 2010).
3.3.3 Lin-28
The small RNA-binding proteins Lin-28 was originally identified as a developmental regulator in Caenorhabidits elegans and is conserved in multiple species (Moss et al. 1997; Moss and Tang 2003; Thornton et al. 2012). Lin-28 suppresses the biogenesis of let-7 by binding to let-7 precursor and facilitating its terminal uridylation and degradation (Thornton et al. 2012; Hagan et al. 2009; Heo et al. 2009, 2012). Lin-28 is up-regulated during myogenesis and promotes myocyte differentiation. In mouse myoblast C2C12 cells, Lin-28 binds to IGF-II mRNA and increases its translation via enhancing its association with RNA-containing translation initiation complexes including eIF3β (Polesskaya et al. 2007).
3.3.4 IMPs
IGF-II mRNA binding proteins 1, 2, and 3 (IGF2BP1; IMP1, IGF2BP2; IMP2, IGFBP3; IMP3) belong to a conserved RNA binding protein family and function in various cellular processes, such as cell polarization, migration, proliferation, differentiation, embryogenesis and metabolism (Bell et al. 2013). IMPs bind to target mRNAs and regulate their expression post-transcriptionally via modulating mRNA stability , localization, and translation (Nielsen et al. 1999). IMPs associate at multiple sites with in the 5′ UTR of IGF-II leader-3 mRNA and repress translation during embryonic development (Nielsen et al. 1999). In contrast, IMP3 functions as a translational activator of IGF-II leader-3 mRNA, thereby promoting cell survival and tumor progression in various cell types, including K562 leukemia cells and glioblastoma cells (Liao et al. 2005, 2011; Suvasini et al. 2011). Together these data suggest that IMPs may influence IGF-II expression in a tissue- or cell-type specific manner. IMP2 binds to an internal ribosome entry site of IGF-II mRNA 5′ UTR and the binding of IMP2 is controlled by the mammalian target of rapamycin (mTOR) pathway (Dai et al. 2011). Phosphorylation of IMP2 on Ser162 and Ser164 residues by mTOR enhances the interaction between IMP2 and IGF-II mRNA, promoting translation of the latter (Fig. 2.5). IMP phosphorylation occurs in the mouse embryo as well as in adult tissues including islets of Langerhans, and seems to promote IGF2 expression, thereby regulating fetal growth and glucose homeostasis (Dai et al. 2011).
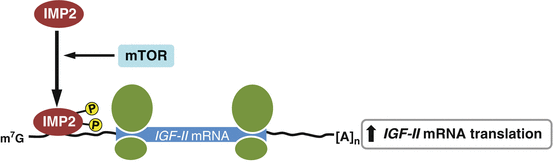
Fig. 2.5
A proposed model of IMP2 action on IGF-II biosynthesis
4 Conclusion
It is clear that the expression of insulin and IGFs is extensively regulated by post-transcriptional processes, including alternative splicing and mRNA stability and translation. Although the factors required for the regulation of INS and IGF mRNA alternative splicing are still largely unknown, the post-transcriptional regulation of INS and IGF mRNA is a complex process involving an array of different trans-acting factors such as RBPs and miRNAs . Here, we have described nearly all trans-acting factors that are known to directly bind to the UTRs of these mRNAs and modulate their expression. In addition, there are numerous factors, not described here, that indirectly regulate their expression at post-transcriptional levels that do not require direct binding to the UTRs. Through the cooperative regulation of both direct and indirect trans-acting factors, the expression of insulin and IGF expression is coordinately regulated. In response to acute changes in circulating glucose and other metabolic stimuli, the rate of insulin biosynthesis rises more dramatically within minutes by rapid increases in the stability and translation of the encoding mRNAs without de novo synthesis of INS mRNA (Brunstedt and Chan 1982; Itoh and Okamoto 1980). A long-term increase in insulin levels is dictated by both de novo synthesis and post-transcriptional regulation of INS mRNA. As described, although IGFs has insulin-like effects in glucose homeostasis, IGFs play a major role in the regulation of cellular processes such as growth, proliferation, and survival, which occur over longer periods. Thus, the IGF levels in these cases are likely changed by the long-term rather than the short-term regulation of the encoding mRNAs.
Related posts:
 Translational and Post-translational Control of Leptin Production by Fat Cells
Translational and Post-translational Control of Leptin Production by Fat Cells
 MicroRNA Regulation of Endocrine Functions in the Ovary
MicroRNA Regulation of Endocrine Functions in the Ovary
 Post-transcriptional and Post-translational Regulation of Steroidogenesis
Post-transcriptional and Post-translational Regulation of Steroidogenesis
 Post-transcriptional Regulation of Prostaglandin Biosynthesis
Post-transcriptional Regulation of Prostaglandin Biosynthesis
 Mechanisms of Post-transcriptional Gene Regulation
Mechanisms of Post-transcriptional Gene Regulation
 Post-transcriptional Regulation of Parathyroid Hormone Gene Expression in Health and Disease
Post-transcriptional Regulation of Parathyroid Hormone Gene Expression in Health and Disease
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


