Fig. 13.1
Mechanisms of GC action discussed in this review. GCs exert their anti-inflammatory action through multiple mechanisms mediating coordinate changes in gene expression (see text). Regulation of inflammatory responses by GC is the result of integrated transcriptional and post-transcriptional pathways. The genes described so far to mediate post-transcriptional control involved in GC action modulate (yellow boxes) inflammatory signaling impacting mRNA turnover and translation, in part by the function of MKP-1, or regulate components that participate directly to the RNP complex conveying changes in mRNA turnover and translation, such as the RNA-binding protein TTP , several miRNAs and their processing enzymes
1.3 Inflammation-Related Genes Targeted by GC through PTR Effects
Alteration of mRNA turnover or translation has been documented for a large number genes involved in inflammation and immunity, such as TNF-α, IL-1α , IL-6, IL-8, interferon-β, granulocyte macrophage colony-stimulating factor (GM-CSF), cyclooxygenase (COX)-2, IL-2, IL-3, inducible nitric oxide synthase (iNOS), vascular endothelial growth factor (VEGF ) and many chemokine genes such as CXCL1, CCL2, CCL7, CCL11, CCL13, and many others (Amano et al. 1993; Tobler et al. 1992; Shaw and Kamen 1986; Peppel et al. 1991; Carballo et al. 2000; Sawaoka et al. 2003; Stoecklin et al. 2003; Ogilvie et al. 2005; Fechir et al. 2005; Fan et al. 2011; Hamilton et al. 2010)—a comprehensive list is in recent reviews (Anderson 2010). The majority of these genes have been demonstrated to be GC-sensitive in many experimental and therapeutic settings and, at the same time, it is known that GC sensitivity can vary in different cell types and can be hampered by inflammatory conditions, leading to GC resistance or at least loss of full therapeutic control. However, the list of GC-regulated genes for which the existence of a PTR component is demonstrated is much shorter (see Table 13.1), though studies utilizing genome-wide approaches are accelerating the pace of the identification of PTR-dependence of GC-mediated gene regulation (Ishmael et al. 2008, 2010). Knowledge of the participation of aberrant PTR response as mechanism of GC resistance is even more ill-defined.
Table 13.1
Genes regulated by glucocorticoids through alteration of mRNA stability : mRNA binding sequences and RBP found to be involved in GC action
Gene | mRNA stability | mRNA region (and element) involved | RBP | References |
|---|---|---|---|---|
COX-2a | Decreased | 3′UTR (ARE) | Unknown | Lasa et al. (2001) |
Cyclin D3 | Decreased | 3′UTR | Unknown | Garcia-Gras et al. (2000) |
CCL2 | Decreased | 3′UTR, 5′UTR (G/C rich region) | TTP, GR | |
CCL7 | Decreased | 3′UTR, 5′UTR (G/C rich region) | TTP, GR | |
CXCL1 | Decreased | 3′UTR | TTP | Ishmael et al. (2008) |
Fibronectin | Increased | Intronic (unspecified) | Unknown | Ehretsmann et al. (1995) |
GM-CSF | Decreased | Unknown | Unknown | Tobler et al. (1992) |
IFNβ | Decreased | ARE | Unknown | Peppel et al. (1991) |
IL-1 | Decreased | Unknown | Unknown | Amano et al. (1993) |
IL-4Rα | Decreased | Unknown | Unknown | Mozo et al. (1998) |
IL-6 | Decreased | 3′UTR | TTP | |
CXCL8 | Decreased | Unknown | Unknown | Tobler et al. (1992) |
iNOS | Decreased | Unknown | Unknown | Korhonen et al. (2002) |
LIF | Decreased | Unknown | Unknown | Grosset et al. (1999) |
SP-1 | Increased | 3′UTR, 7.6S region | Unknown | Huang et al. (2012) |
TNFα | Decreased | ARE | TTP | Smoak and Cidlowski (2006) |
VEGF | Decreased | Unknown | Unknown | Gille et al. (2001) |
2 Post-transcriptional Determinants of GC Anti-inflammatory Action
Mechanistic knowledge on how immune responses rely on PTR for coordinate expression of cytokines, chemokines and other inflammation-related factors is lending a strong rationale to investigate GCs’ ability to target PTR mechanisms as part of their anti-inflammatory activity. Following are studies that have started unraveling the specific PTR determinants—the signaling mediators, specific regulatory functions, RNA binding sequences, RBPs and miRNA functions, etc.—involved and necessary for GC action. Further studies are necessary to fully characterize GC-driven PTR components and to develop experimental systems that could establish and measure the overall impact of PTR in GC’s anti-inflammatory therapeutic action.
2.1 GC Effect on Signaling Pathways Regulating PTR Functions
According to transcript-, stimulus- and cell-specific-mechanisms, multiple signaling pathways can regulate—through different post-translational modifications—the trafficking, binding affinity, compartmentalization and other functional aspects of RNA-binding proteins and miRNAs . Members of the mitogen-activated protein kinases (MAPK) family, such as the stress -activated protein kinase/c-Jun N-terminal kinase (SAPK/JNK), p38/SAPK2 and extracellular regulated kinase (ERK) are essential—though obviously not exclusive—regulators of PTR, in full integration with their upstream control on gene transcription (Kracht and Saklatvala 2002; Gaestel 2006). Post-transcriptional activities of MAP kinases have been increasingly well-characterized in mechanistic terms and recognized as necessary for inflammation-driven overexpression of a large number of inflammatory targets (Kracht and Saklatvala 2002; Dean et al. 2004; Anderson 2008; Gaestel 2013). At the same time, inhibition of MAPK signaling constitutes a major and powerful component of GC anti-inflammatory action, better characterized with regard to the MAPK signaling control of transcriptional mechanism (Kracht and Saklatvala 2002; Clark et al. 2003; Clark and Lasa 2003). Although MAPK-dependent transcript stabilization by inflammatory stimulation has been demonstrated for many transcripts that are described as susceptible to GC inhibition through acceleration of mRNA decay —among these IL-1, VEGF , COX-2, TNFα, IL-6 and IL-8 and many chemokines (Amano et al. 1993; Gille et al. 2001; Lasa et al. 2001; Swantek et al. 1997; Tobler et al. 1992; Ishmael et al. 2008; Stellato 2004), relatively few studies to date have directly investigated the PTR regulatory sequences and binding partners targeted by GC as part of the inhibitory effect on MAPK-dependent gene regulation , and attempt to measure the overall anti-inflammatory impact of this regulatory mode. Such experimental evidence would be relevant for identifying specific pathways potentially targetable for a selective anti-inflammatory therapeutic intervention. The most relevant studies on GC effect to this end have focused, in several experimental models, on p38-regulated PTR showing that treatment with specific p38 inhibitors in activated cells antagonized kinase-induced mRNA stabilization (or increased translation) and promoted acceleration of mRNA decay or decreased translation, effects that were found to be mediated by the ARE -bearing 3′-UTRs of these genes (Kracht and Saklatvala 2002; Zhang et al. 2002; Dean et al. 2004). One of the first studies to characterize the involvement of p38 MAPK- mediated PTR by GC action used COX-2 mRNA turnover as model (Lasa et al. 2000, 2001). The decay of a β-globin reporter including the ARE-bearing 3′-UTR of COX-2 mRNA was transiently transfected in HeLA cells and examined upon stimulation by a constitutively active form of MAPK kinase-6 (MKK-6), an upstream activator of p38, which increased the reporter’s mRNA stability . This effect was inhibited both by cell pretreatment with dexamethasone (Lasa et al. 2001) and p38 antagonists (Lasa et al. 2000). As in multiple systems (Clark and Lasa 2003), GC cell treatment inhibited stimulus-induced p38 phosphorylation, supporting the conclusion that GC effect on the reporter mRNA turnover occurred through the inhibition of p38 MAPK activity, in turn responsible for COX-2 mRNA stabilization (Lasa et al. 2001).
The effect of GC on MAPK-driven PTR control—that mediated by p38 in particular—was later found to be significantly linked to GC-induced transcriptional activation of the MAPK phosphatase 1 gene (MKP-1), which antagonized the p38-mediated stabilization of COX2 mRNA (Lasa et al. 2002). GC-driven MKP-1 induction was confirmed in several cell types including macrophages and airway epithelium (Imasato et al. 2002). Earlier data showed that GC effects on mRNA stability of COX-2 and other inflammatory transcripts did require de novo gene expression (Newton et al. 1998), supporting the hypothesis that MKP-1 contributed to the post-transcriptional mechanisms of GC action observed for COX-2 regulation. More recent studies using either transgenic or KO models of MKP-1 gene have shown its non-redundant role as negative regulator of inflammation and as important factor of GC anti-inflammatory mechanism, as suggested by the significant impairment of GC response in macrophages of MKP-1−/− mice (Abraham and Clark 2006; Liu et al. 2007). Recently, overexpression or ablation of MKP-1 in LPS-stimulated rat macrophages was found to regulate levels of IL-6, IL-10 and TNFα mRNA through changes in mRNA stability in conjunction with regulation of nucleocytoplasmic shuttling of AUF-1, the ARE -binding RNP promoting the mRNA decay of those cytokines (Yu et al. 2011) and, as discussed earlier, shown in KO model to be involved in control of TNF-α, chemokine and Th2-driven inflammation (Sadri and Schneider 2009). Studies on GC effects in similar systems will be necessary to identify the specific molecular species downstream of MAPK through which GC exert PTR control of inflammatory signaling—and how much of it is dependent on MKP-1—in order to dampen the inflammatory process.
The PTR effects by GC documented to be ARE -dependent and mediated by MAP kinases indicate that such mechanism can be cell-specific (Lasa et al. 2001, 2002) and, possibly, stimulus- and transcript-specific. As not all ARE-containing genes are p38-dependent (Tebo et al. 2003; Frevel et al. 2003), the signaling pathways conveying ARE-dependent, GC-driven PTR requires further scrutiny. It is clear, in fact, that additional mechanisms of control by GC on PTR signaling may very well exist outside the p38-MKP1 axis. For example, dexamethasone inhibited cytokine-induced IL-8 in human airway epithelial cell lines by acceleration of mRNA decay , in parallel with induction of MKP-1 expression; however the effect on IL-8 mRNA stability was not recapitulated by overexpression of MKP-1 nor influenced in opposite way by its silencing, suggesting that the post-transcriptional effect of GC on IL-8 expression can be conveyed by molecular species downstream of MKP-1 (Dauletbaev et al. 2011). In particular, the effect of GC on MAPK-activated protein kinases (MAPKAP) 2 and 3, which are master regulators of mRNA decay and translation through phosphorylation of TTP and other major RBPs deserves further experimental testing (Moens et al. 2013; Gaestel 2013).
2.2 Sequence Elements for Regulations and RNA-Binding Proteins Conveying GC Action
Genes required for a specific biological process—a specific pool of transcription factors, cytokines, chemokines, enzymes required for a TLR-mediated activation, for example—can be regulated post-transcriptionally in a coordinate fashion via a specific RBP that binds to common regulatory sequences present in those mRNAs, as a post-transcriptional regulon (Anderson 2010; Keene 2007). Therefore, control of expression or function of the RNP components by GC may convey a large, yet rather specific impact on gene networks involved in specific pathophysiological processes, by acting on a regulatory step well downstream of transcription. In keeping with the immunomodulatory function of GC action, it can be expected however that components of post-transcriptional gene control may change in sensitivity to GC according to tissue- and cell-specific issues, and according to the type of inflammatory event and the ensuing signaling. Characterization of GC action on the components of the RNP complex—the RNA regulatory sequences and their binding partners—is a growing area of investigation aimed at identifying more specific anti-inflammatory strategies.
GC and Sequence Elements for Regulations
The interaction of regulatory factors—RNA-binding proteins, microRNAs and potentially other sncRNAs—with sets of relatively specific sequences present on the targeted mRNA is at the core of post-transcriptional gene regulation (Bakheet et al. 2006). The targeted sequences—heterogeneous in sequence, secondary structure, position and specific functions—are more frequently represented in the 5′ and 3′ untranslated regions (UTR ) of the mature cytoplasmic mRNA, and have been collectively denominated USER (Untranslated Sequence Elements for Regulations) (Wilusz and Wilusz 2004; Keene 2007). The majority of inflammation-driven, MAPK-dependent PTR as well as many of the described post-transcriptional effects by GC occur through association of regulatory factors to adenylate/uridylate-rich elements (AREs) within the 3′-UTR of mRNAs. This evolutionarily conserved, heterogeneous group of sequences have been the first to be described as central in PTR events (Shaw and Kamen 1986) and in immune-mediated PTR processes (Chen and Shyu 1995; Anderson 2010), as indicated by the fact that immune-related genes are highly enriched in AREs (see http://rc.kfshrc.edu.sa/ared, http://rna.tbi.univie.ac.at/AREsite) (Bakheet et al. 2006; Gruber et al. 2011). In one of the first demonstrations of ARE -dependent GC effect, macrophages from mice expressing the TNF-α gene carrying the deletion of the ARE (ΔARE) overexpressed TNF-α protein due to decreased decay rate of TNF-α mRNA as well as to loss of translational inhibition (Kontoyiannis et al. 1999). In these cells, LPS-induced increase in TNF-α became unresponsive to the inhibitory effect that a p38 inhibitor displayed on WT cells, showing dependence of p38/JNK-mediated activation of TNF-α translation. Conversely, cell treatment with dexamethasone of ΔARE macrophages retained in part its ability to inhibit TNF-α secretion, suggesting that the ARE-dependent inhibition of TNF-α is an important—but not exclusively p38/JNK-mediated- PTR mechanism of inhibition of TNF-α by GC. There are currently no data on GC regulation on ARE-BP acting as repressors of TNF-α translation, such as T-cell intracellular antigen (TIA) and TIA-related protein (TIAR) (Zhang et al. 2002). As further experimental proof of ARE-dependent GC action, the dexamethasone-induced acceleration of an mRNA reporter expressed by a construct containing COX-2 3′UTR was suppressed by deletion of a small ARE cluster within the 3′-UTR, which bears a large number of AREs (Lasa et al. 2001). Dependence from 3′UTR of GC action on mRNA stability , though not specifically addressing ARE-dependence, has been described for several inflammatory genes (Stellato 2004) (see Table 13.1). No data are so far available on the participation to GC action of other classes of 3′UTR USER, such as the GU-rich elements (Halees et al. 2011) and is so far unknown whether endogenous GC would support PTR mechanisms keeping inflammatory transcripts highly unstable at baseline conditions, as documented through binding of the RBP Roquin to the constitutive decay element (CDE) present as well in the 3′UTR for TNFα mRNA and other transcripts (Leppek et al. 2013). Other sequences outside the 3′UTR have been found to mediate the effect of GC on mRNA turnover and/or translation. In particular, GC-mediated PTR has been recently linked to sequences present in the 5′UTR of CCL2 mRNA and other transcripts. Initial data generated in rat smooth muscle cells (SMC) indicated that the increase in decay rate of CCL2/MCP-1 mRNA induced by cell incubation with GC was not dependent on the ARE-bearing 3′-UTR but was rather mediated by a unique sequence in the 5′-UTR (Poon et al. 1999). Recently, GC-induced increased decay of CCL2 mRNA in human airway epithelial cells has been ascribed to a Guanidine/Cytidine (GC)-rich motif identified in the 5′UTR region in the CCL2 mRNA (Ishmael et al. 2010). This sequence mediated the transcript association of a complex containing the GC receptor (GR), which operated in this experimental setting as a bona-fide RNA-binding protein (see next paragraph).
An important non-immune gene induced by GC in alveolar type 2 epithelial cells encodes the surfactant protein B (SP-B), necessary for appropriate function of the surfactant fluid lining the alveolar space in the lungs. Together with transcriptional control, GC increase SP-B mRNA stabilization by a 3′UTR -dependent mechanism requiring a 126-nt-long segment denominated 7.6S region (Huang et al. 2008). Mutagenesis studies indicated that within this region, a 30-long element predicted to form a stem-loop structure was sufficient for GC-mediated mRNA stabilization (Huang et al. 2012).
Considering the number of regulatory sequence elements within 5′-UTR and 3′-UTR of eukaryotic mRNAs besides ARE (see http://utrdb.ba.itb.cnr.it/) (Grillo et al. 2010), the interactions that could occur among the regulatory factors assembling over multiple USERs present in the same transcript, together with specific changes brought to the RNP complexes by stimulus-dependent signaling, it is evident that much more remains to be discovered on the identity and characteristic of additional USER supporting GC action and their binding partners.
RBPs as Targets/Mediators of GC Function
RNA-binding proteins are among the most abundant and well-conserved eukaryotic genes (Gerstberger et al. 2014). They regulate several aspects of the mRNA metabolism, such as its maturation, nucleocytoplasmic transport, subcellular localization, rate of decay, and translation (Dreyfuss et al. 2002; Anderson 2008, 2010) and their key regulatory roles in immune responses is now well established (Ivanov and Anderson 2013; Yuan and Muljo 2013). Target-bound RBPs implement their functions by forming dynamic multimeric complexes with miRNAs , other RBPs and RNA-degrading enzymatic proteins not directly interacting with the mRNA (Jiang and Coller 2012; Ciafre and Galardi 2013; Srikantan et al. 2012; Steitz and Vasudevan 2009), often co-localized in cytoplasmic foci of mRNA metabolism (P bodies and stress granules) (Anderson and Kedersha 2009; Bhattacharyya et al. 2006; Jing et al. 2005). Numerous ARE ‐binding proteins (ARE‐BPs) have been functionally characterized as regulatory factors of mRNA decay and translation of inflammatory and immune genes [for review see (Dreyfuss et al. 2002; Stoecklin and Anderson 2006a; Anderson 2008, 2010)], but very few have been investigated with regard to their potential function in GC action. While numerous proteins—among them TIA‐1, TIAR, FXR1P, CUGBP2 —can exert translational repression, changes in mRNA stability leading to prolonged half-life and increased expression of a vast number of inflammatory genes have been ascribed mostly to HuR , the ubiquitous member of the Hu family of RBPs (Brennan and Steitz 2001; Meisner and Filipowicz 2011; Simone and Keene 2013) and to YB1, described for a more limited number of transcripts, among which IL-2 mRNA (Chen et al. 2000; Lyabin et al. 2014). In contrast, numerous RBPs have been characterized as involved in destabilizing ARE‐bearing inflammatory mRNAs, such as AUF1 , KSRP , RHAU, and members of the TTP /BRF family of proteins (for review see Stoecklin and Anderson 2006b). Despite their documented role as regulatory factors for established GC-sensitive genes—cytokines, chemokines and enzymes involved in inflammation—as well as their strong functional dependence from MAPK signaling pathways (Sugiura et al. 2011) that are as well GC-sensitive, the biology of RNA-binding proteins in human inflammatory diseases is presently ill-defined and knowledge on the mechanisms of control by GC, or by anti-inflammatory intervention in general, on their expression and function is lagging behind in translational research settings. Following are the main observations of studies that have so far addressed directly the role of RBPs in GC-mediated PTR.
Tristetraprolin: A Mediator of Post-transcriptional GC Action
Tristetraprolin (TTP ) is the product of the ZFP-36 gene (also known as TIS11, Nup475, and GOS24) and member of a family of CCCH zinc finger proteins that include TTP, Butyrate-response factor (BRF)-1 and BRF-2 (Carrick et al. 2004). These proteins mediate deadenylation -dependent mRNA decay (Raineri et al. 2004) through binding of zinc finger domains to adjacent UUAU/UUAU half-sites, which trigger complex interactions with components of the exosome, the decapping /Xrn1 complex, and with the RISC complex (Worthington et al. 2002; Blackshear et al. 2003a; Brewer et al. 2004; Stoecklin and Anderson 2006b). Tristetraprolin is induced as an immediate early response gene by inflammatory mediators, phorbol esters, LPS and growth factors in a number of immune and structural cell types, including T cells, macrophages and fibroblasts where it displays a predominantly cytoplasmic localization (Blackshear 2002). TTP is also expressed in lung, liver, and intestine tissues (Cao 2004; Lu and Schneider 2004). As previously mentioned, the importance of TTP as endogenous rate-limiting factor of the inflammatory response has been convincingly demonstrated in TTP−/− mice, which develop severe inflammatory arthritis, autoimmune dysfunction and myeloid hyperplasia through the deregulated expression of TNF-α and GM-CSF (Taylor et al. 1996). Tumor necrosis factor α is both a major target and regulator of TTP, as TNF-α induces TTP synthesis, which in turn leads to destabilization of TNF-α mRNA (Carballo et al. 1998). TTP also modulates mRNA decay rates of GM-CSF, COX-2, INOS, IL-2, IL-3, IL-10, and several chemokines, such as CCL2, CCL7, CXCL1, CXCL8 and others (Lai et al. 2006; Carballo et al. 2000; Sawaoka et al. 2003; Stoecklin et al. 2003; Ogilvie et al. 2005; Fechir et al. 2005; Ishmael et al. 2008). Additional transcripts whose decay is regulated by TTP have been identified in a recent genome-wide study of mouse embryonic fibroblasts (MEFs) isolated from TTP-knockout (TTP−/−) mice (Lai et al. 2006), and in mouse macrophages in which TTP expression was silenced (Jalonen et al. 2006). Importantly, phosphorylation of TTP by p38‐MAPK and the downstream MAPKAP, MK2 (Carballo et al. 2001; Mahtani et al. 2001) is an important mechanism of post-transcriptional control during inflammation, as it leads to functional inactivation of TTP due to an inhibitory complex with the adaptor protein 14‐3‐3 (Chrestensen et al. 2004; Johnson et al. 2002; Stoecklin et al. 2004), though this could be dependent by contextual stimuli (Rigby et al. 2005). Furthermore, TTP has a key role as MK2/3-regulated RBP in limiting TNFα translation (reviewed in Gaestel 2006, 2013). Overall, the function, regulation and targeted genes of TTP make this molecule an ideal mediator of GC anti-inflammatory activity.
Indeed, expression of TTP and BRF-1 has been reported to be induced by GC in primary human airway epithelial cells and epithelial cell lines (Pawliczak et al. 2005; Ishmael et al. 2007). Induction of TTP by GC has been shown in vivo by treatment with dexamethasone in adrenalectomized rats, which caused a three- to fourfold increase of TTP mRNA expression in the lung, liver and thymus (Smoak and Cidlowski 2006). Based on these results, the mechanism of TTP induction by GC was further defined using the airway human epithelial cell line A549. An important characteristic of TTP regulation by GC found in this system is the extended length of induction of TTP mRNA, which was detectable up to 8 h following GC stimulation. This contrasts with the rapid but transient induction by TNFα, where TTP mRNA peaked after 2 h and returned to baseline within 4 h from stimulation. At the protein level, both TTP and TNFα sustained in A549 cells a prolonged expression of TTP up to 24 h post-stimulation. Concordance of the action on TTP expression by TNFα and GC may appear counterintuitive, yet it can be reconciled viewing TTP as a molecule involved in homeostatic limitation of an acute inflammation, a process which ‘begins with the end in mind’ (Anderson 2010), with induction of TTP being advantageous as a negative feedback mechanism in both settings. From a mechanistic standpoint, GC-induced TTP expression was found to be transcription—and GR binding—dependent, as it was abolished by cell pretreatment with the transcriptional inhibitor actinomycin D and by GR antagonist RU486, respectively; paired with experiments showing no change in TTP mRNA stability , run-on experiments confirmed that TTP induction by GC was driven in A549 cells by increased transcription, though a bona fide GRE was not identifiable from the published hTTP sequence. Chromatin immunoprecipitation (ChIP) experiments in GC-treated cells showed enrichment of GR recruitment in two regions of the TTP gene: a 5′ flanking portion of the promoter containing binding sites for STAT, Smad, and NF-κB—all transcription factors known to engage protein–protein interaction with GR—and a portion of the 3′ flanking region which contains a half-GRE sequence. Silencing of TTP expression in A549 cells significantly impaired inhibition of its major target, TNF-α by GC, demonstrating a functional role of GC-induced TTP. This process was dependent on the TNF-α 3′UTR , as GC treatment selectively decreased, in transfected A549 cells, the expression of a luciferase reporter carrying the TNF-α 3′UTR. These results indicate for the first time that GC inhibition of TNF-α is mediated by 3′UTR-mediated post-transcriptional mechanisms critically dependent by induction of TTP. However, additional tissue- and/or stimulus-specific factors regulating GC influence on TTP expression are also suggested by a study reporting that LPS-induced TTP expression is instead inhibited by GC in mouse macrophages (Jalonen et al. 2005).
The work by Ishmael et al. (2008) extended GC sensitivity of TTP expression also to human primary bronchial epithelial cells and examined the role of TTP in GC anti-inflammatory action on a genome-wide scale. Mouse embryonic fibroblasts (MEFs) cell lines developed from TTP KO mice, in comparison to MEFs of wild-type (WT) littermates were treated the topical GC budesonide or diluent control for 3 h and then treated in the absence or presence of 10 ng/ml TNF-α for 1 h. The number of genes regulated in WT cells was largely reduced in TTP−/− cells, with the greatest loss of response (97 %) accounted for genes that in WT were down-regulated by GCs: only 5 of the 145 GC-responsive genes in WT were still comparably repressed in TTP−/− cells (Fig. 13.2a). Interestingly, also GC-induced genes were diminished in TTP−/− cells by 89 % compared to WT cells, suggesting a vast network of indirect effects. TNF-α-dependent responses were also globally affected in the absence of GC treatment, pointing again to the role of TTP in limiting the amplitude of acute inflammation either in response to triggering stimuli or as part of the endogenous anti-inflammatory mechanisms. Of particular relevance, the chemokines CCL2, CCL7, CXCL1, CXCL5 and CXCL7 were among the genes whose inhibitory response to GC was significantly lost in TTP−/− MEF. Such targeting reinforces the importance of TTP-mediated function in the anti-inflammatory mechanism of GC action: chemokines are in fact key determinants of recruitment and activation of inflammatory cells within the inflamed mucosa and as such, they are main targets of GC therapy in chronic inflammatory diseases such as rhinosinusitis, asthma and COPD (Stellato 2007; Stellato and Schleimer 2000). Genome ontology analysis confirmed that regulated genes were functionally enriched in immunity and signal transduction, processes that are chiefly targeted by GCs for their anti-inflammatory action (Fig. 13.2b). Involvement of TTP also on genes important for GC control of metabolism and cell development indicate that TTP is likely to mediate other homeostatic and nonimmune functions driven by GCs. Loss of GC response was validated in the same study for selected targets in WT MEFS, as well as in primary human epithelial cells (unpublished data, C. Stellato), following transient TTP silencing, validating the general hypothesis beyond the TTP−/− mouse model.
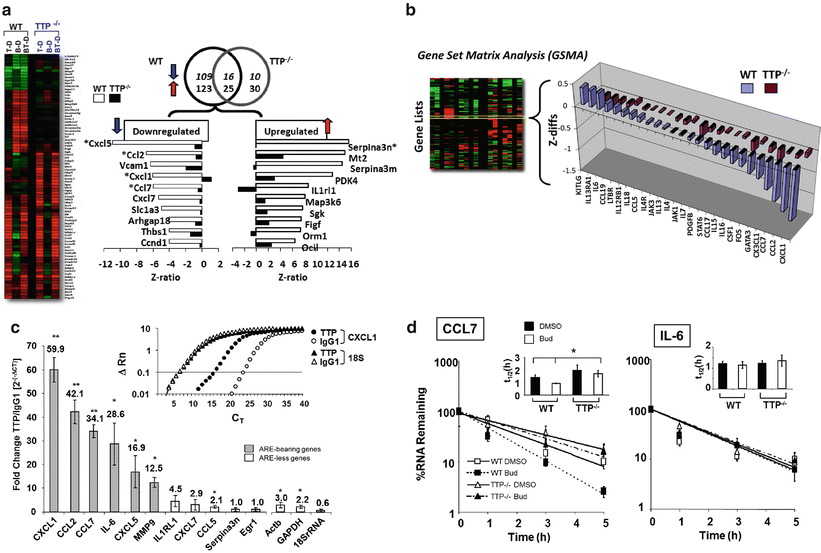
Fig. 13.2
GC-mediated gene regulation is significantly altered in TTP −/− murine fibroblasts. (a) Differential regulation of gene expression in WT and TTP−/− MEFs. Left, Heat maps of the array data sets from WT or TTP−/− cells and right, changes in GC response for selected genes (expressed as Z ratio) in WT vs TTP−/− cells upon exposure to budesonide (Bud) versus diluent-treated cells. (b) Genome ontology analysis by Gene Set Matrix Analysis (GSMA) showing loss of GC response for genes involved in asthma pathogenesis in TTP−/− MEFs. The Gene Set Matrix Analysis (GSMA) algorithm allows the identification of coordinated changes in gene expression. Experimental conditions are examined against “gene lists” pooled according to a biologically relevant grouping system instead of against each gene present on the array. (c) Association of TTP with GC-regulated mRNA targets identified by immunoprecipitation of endogenous mRNA-TTP complexes. Upper right, Representative real-time PCR amplification plot of fluorescence intensity over background (ΔRn) against PCR cycle (CT) showing enrichment of CXCL1, but not of 18S RNA transcripts, in lysates of GC-treated MEFs subjected to IP with an anti-TTP Ab (filled symbols) vs the IP with the isotype-matched Ab (open symbols). Bargraph, enrichment of immunoprecipitated transcripts by the anti-TTP relative to IP with isotype Ab control (fold values indicated). Of the 11 transcripts tested, 6 (grey bars) contain multiple AREs. (d) Differential effect of TTP loss in GC-induced CCL7 and IL-6 mRNA decay . Results are shown as the percent of mRNA at time 0 in actinomycin-treated cells. The bar graph insets indicate the half-life (t (1/2), in h) in each condition. Modified from Ishmael et al. (2008), reprinted with permission
Probing the mechanism of TTP function in GC response revealed a high level of complexity involving direct, ARE -mediated effects as well as indirect effects. For a group of 11 transcripts selected among those that lost their GC sensitivity in TTP−/− cells, those bearing TTP- compatible sequences such as AUUUA pentamers or UUAUUUAUU nonamers (Stoecklin et al. 2008)—CCL2, CCL7, CXCL1, CXCL5 as well as IL-6 and MMP9—showed association with TTP, documented through selective immunoprecipitation of ribonucleoprotein complexes (RNP-IP) using anti-TTP ab and subsequent real-time PCR; the transcripts showing no association with TTP by RNP-IP, despite profound changes in GC sensitivity in TTP−/− cells—IL1RL1, CXCL7, CCL5, Serpin3n, Egr1—presented instead only one or no AREs in the 3′-UTRs , with the exception of EGR-1 mRNA, which bears two UAUUUAU heptamers in an A/U-rich setting (Fig. 13.2c). However, among the transcripts found to be associated to TTP, GC-dependent changes in mRNA decay rate in TTP−/− cells—assessed by actinomycin D treatment—were heterogeneous, challenging the assumption that TTP would mediate GC action exclusively by accelerating ARE-mediated mRNA decay (Fig. 13.2d). For CCL7 and CCL2 mRNA, the acceleration of mRNA decay induced by GC in WT MEFs was no longer present in TTP−/− cells, in line with the more straightforward hypothesis. On the contrary, IL-6 mRNA decay was not modified by either GC treatment or by lack of TTP, despite significant mRNA enrichment in the RNP-IP assay and the presence of a 3′-UTR extremely rich in AREs. CXCL5 mRNA became unstable in TTP−/− cells compared to WT cells already in the absence of GC, indicating that TTP supported in this case a relative stabilization of CXCL5 basal mRNA turnover rate and that the inhibitory effect of GC seen in TTP−/− cells was likely a consequence to the loss of this function, rather than the GC action in itself. Further remarking the complexity of TTP biology—and possibly also the limitation of the Actinomycin D-based experimental system used in the study—these data implicate that besides ARE-mediated mRNA decay, TTP could form RNPs through protein interaction with other RBPs (Anderson 2008) and/or by establishing cooperative or antagonistic function with miRNAs (Jing et al. 2005; van Kouwenhove et al. 2011) (see following dedicated paragraph). Prototypic of this context-driven, rather than strictly ARE-dependent, function is how TTP promote stabilization of the ARE-rich INOS mRNA by interacting with the ARE-bound KH-type splicing regulatory protein (KSRP ). Under proinflammatory stimulation, TTP association with KSRP would remove this mRNA decay-inducing factor and the associated exosome and allow binding for the RBP HuR , resulting in INOS mRNA stabilization (Fechir et al. 2005). Many of the transcripts found to be up-regulated upon GCs treatment in TTP−/− cells could be similarly susceptible of TTP regulation through critical changes within the regulatory RNP complexes. In addition, GC could modulate TTP function also through changes in TTP phosphorylation, thus affecting functional aspects (change its localization, function or affinity for its binding site) rather than solely its synthesis.
TTP -mediated effect of GC can indirectly affect a larger gene pool when targeting a regulatory protein: an example from the study by Ishmael is the TTP-dependence of GC inhibition of the transcription factor Egr-1, which would also affect downstream expression of Egr-1-dependent proinflammatory genes (Cho et al. 2006; Ingram et al. 2006) underscoring how regulation of mRNA turnover can indirectly affect transcriptional control of multiple genes.
The effect and mechanisms of GC action on TTP -mediated translational control of gene expression, whose importance is documented for TNFα, remains to be explored as for that exerted on other RBPs known to regulate translation—such as TIA, TIAR and others (Gaestel 2013).
Glucocorticoid -Receptor (GR)-Mediated Chemokine Decay: A Novel Mechanism of GC-Mediated PTR
The glucocorticoid receptor (GR) is a steroid hormone receptor belonging to the superfamily of nuclear receptors (NR), which exert gene regulatory functions as transcription factors (Gronemeyer et al. 2004). Transcription factors, such as the GR, and RBPs share important functional characteristics, such as sequence-specific interaction with nucleic acids, activation-dependent nucleocytoplasmic shuttling, and the ability to affect multiple targets through binding to conserved nucleic acid sequences. Taking this similarity even further, some protein factors can bind to both DNA and RNA—as in the case of the zinc-finger protein NF-90 that regulates IL-2 transcription through binding to the NFAT site (Shi et al. 2007) as well as mRNA turnover and translation of a vast group of transcripts through AU-rich-specific interactions (Kim et al. 2007). Other two zinc-finger proteins, Yin-Yang (YY)1 and TTP , as well as nucleolin participate in both nuclear and cytoplasmic regulatory events by ribonucleoprotein- and protein–protein interactions either with DNA and transcription factors or with RNA, miRNA and RBPs in the cytoplasm (Chen et al. 2000; Singh et al. 2004; Mongelard and Bouvet 2007; Liang et al. 2009; Stoecklin et al. 2008).
Whether the GR may have a similar ability was hypothesized based on the growing data on PTR contribution to GC action (Stellato 2004). The specific role of GR in mRNA stability was tested for the first time in a model of rat aortic smooth muscle cells (SMC) (Dhawan et al. 2007), based on earlier observation by the same group of the ability of the synthetic GC Dexamethasone (Dex) to accelerate mRNA decay for CCL2/MCP-1 mRNA (Poon et al. 1999). Experiments using RNA electromobility shift assay (R-EMSA) showed an upward mobility shift of the complex formed by cytoplasmic extracts of Dex-treated SMC after a short incubation (5 min) with radiolabeled full length CCL2/MCP1 mRNA compared to control cytoplasmic extracts, suggesting that additional proteins bind to the transcript under Dex control. Using a cell-free mRNA stability assay, in which degradation of a labeled synthetic CCL2/MCP-1 transcript was detected over time following incubation with cytoplasmic SMC lysates, the authors showed that incubation for 30 min of the Dex-treated SMC extracts with the CCL2/MCP-1 probe led to its complete degradation, while the extracts from control SMC did not change its detection; strikingly, the probe degradation was specifically inhibited by preincubation of Dex-treated SMC extracts with multiple anti-GR antibodies (Fig. 13.3a). Direct GR-mRNA interaction was confirmed by R-EMSA using recombinant human GR and an in vitro transcribed, human CCL2/MCP-1 mRNA probe (Fig. 13.3b). This interaction was hormone receptor—and mRNA—specific, as neither incubation with other NRs or usage of three alternative transcripts reproduced the results obtained with GR-CCL2 coupling. It is also noteworthy that the shift observed with extracts from Dex-treated cells was higher than that obtained by rhGR alone, suggesting that in intact cells, GR is likely part of a multimeric RNP complex; this was further suggested by the ability of recombinant GR to inhibit transcript degradation by Dex-treated extracts and by enrichment of MCP-1 mRNA detected by immunoprecipitation of RNP complexes (RNP-IP) with an anti-GR specific antibody. These data indicated a previously unknown cytoplasmic function for the GR as well as the existence of novel regulatory sequences, in addition to the conserved ARE , as determinants of CCL2 mRNA stability.
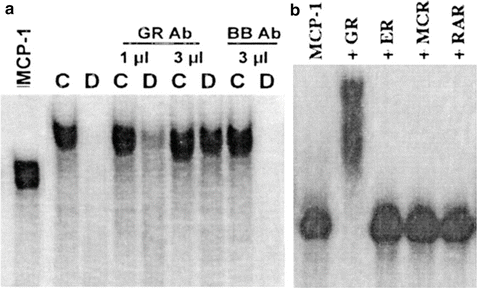
Fig. 13.3
The Glucocorticoid Receptor (GR) associates with MCP-1/CCL2 mRNA and promotes its degradation. Rat aortic endothelial cells were treated with diluent control (C)/dex (D) for 3 h and cytoplasmic lysates was incubated with full length, in vitro transcribed CCL2/MCP-1 mRNA for 30 min ± GR Ab for EMSA essay. (a) Incubation with control cell lysates (C) shows a probe shift indicating formation of an RNP complex; CCL2 probe is degraded in dex-treated cells (D), an effect prevented specifically and concentration-dependently by lysate preincubation with an anti-GR ab. (b) Direct binding of GR with in vitro transcribed CCL2/MCP-1 mRNA was demonstrated by complex formation following probe incubation with recombinant GR; the interaction was specific with respect to other nuclear receptors. Modified from Dhawan et al. (2007), reprinted with permission
The study by Ishmael et al. (2010) aimed to study whether this novel mechanism was in place in a human cellular model of GC response and tested the hypothesis that in functioning with an RBP-like function, GR-mediated control of mRNA decay would have a more global role in GC-mediated PTR action. First, R-EMSA experiments conducted with cytoplasmic extracts from human primary airway epithelial cells and the epithelial cell line BEAS-2B indicated that biotin-labeled CCL2 and CCL7 mRNA, but not CCL5, associated with GR upon cell treatment with the synthetic GC budesonide and were degraded by GC treatment, an outcome prevented by GR neutralization by a specific anti-GR ab in epithelial cell lysates. The association was validated by RNP-IP (Helou et al. 2008) in which cytoplasmic lysates from control and GC-treated epithelial cells were incubated with anti-GR antibody—or with an IgG isotype control to measure background binding—to immunoprecipitate endogenous GR-mRNA complexes, and identified bound CCL2 and CCL7 by PCR in GC-treated samples, similarly to what the same group reported for chemokine transcripts interaction with TTP (Ishmael et al. 2008). Biotin pulldown experiments probed for the binding site of GR using truncated transcripts containing the 5′UTR , coding region (CR) and 3′UTR sections (Fig. 13.4a). In the study of GC-induced decay of rat CCL2 mRNA, transcript stability was not dependent from the ARE or other sequences in the 3′UTR but relied instead on the presence of three stem loops in the transcript 5′-UTR (Poon et al. 1999). As in the rat CCL2 system, association of GR was observed predominantly with the 5′UTR construct, with minimal signal with the 3′UTR construct. The binding site was further mapped through cross-linking of purified recombinant GR with truncated 5′UTR regions (indicating actual nucleic acid-protein interaction) and ultimately localized to a 15-mer fragment localized between nucleotide 44 and 60 (Fig. 13.4b).
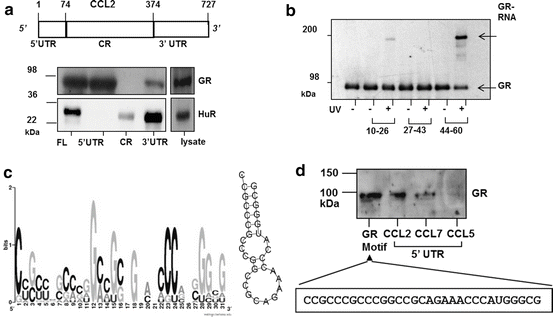
Fig. 13.4
The GR binds to CCL2 mRNA and to multiple targets in human epithelial cells via a G/C-rich Motif present in the 5′ UTR . (a) Top: schematic of CCL2 mRNA, with nucleotide numbers corresponding to untranslated regions (UTR) and coding region (CR). Below, detection of GR by Western blot by biotin pulldown assay from unstimulated BEAS-2B cell lysate, using biotinylated CCL2 full-length RNA (FL), the 5′ and 3′ UTRs , or the CR. The RBP HuR is detected as positive control for association with CCL2 3′UTR. (b). UV cross-linking of purified GR protein to 15-nt fragments of the CCL2 5′UTR, followed by detection by Western blot of the covalently-linked GR via mobility shift of the RNA-bound GR protein (GR-RNA). (c) Left, graphic logo representing the probability matrix of the GR motif, showing the relative nucleotide frequency for each position within the motif sequence. Right, secondary structure of the GR motif comprising the nucleotides with highest frequency for each position within the motif shown in A. (d) Biotin pull-down assay showing association of recombinant GR from unstimulated BEAS-2B cell lysates with the GR motif shown in (c) (sequence shown below), compared to GR association with the full-length 5′UTR of CCL2, CCL7 and CCL5 mRNA (the latter as negative control). Modified from Ishmael et al. (2010); Panganiban et al. (2014), reprinted with permission
As for transcription factors, RBPs interact with nucleic acids over specific cis-regulatory sequences to implement common regulatory function over multiple genes. Therefore, validation of GR function as an RBP would entail the ability to associate with multiple transcripts through a specific sequence and regulate their decay/translation rate. This paradigm was tested in the human airway epithelial cell line BEAS-2B employing RIP-ChIP analysis in which transcripts bound to the endogenous GR were identified by gene arrays after specific IP (Tenenbaum et al. 2002; Keene et al. 2006). Statistically significant mRNA enrichment in GR-IP arrays versus IgG control-IP arrays was found for approximately 500 transcripts, supporting the hypothesis of a broader RBP function for the GR besides interaction with CCL2 and CCL7 mRNAs. The common sequence motif shared by the transcripts associated with the GR, searched using a specific algorithm for RNA sequences (Lopez de Silanes et al. 2004; Kim et al. 2007; Kuwano et al. 2010), consisted in a Guanidine/Cytidine (GC)-rich sequence (78 % of the signature motif), spanning 27–31 nucleotides and forming two contiguous loops on a short stem (Fig. 13.4c), that was proven to bind to recombinant GR via biotin pulldown assay. The entire UniGene database was queried based on this motif and 7889 transcripts displayed a compatible sequence in their 5′UTR (45 % of the entire database) (Fig. 13.4d).
Collectively, the data from these three studies convincingly indicate that the GR can mediate GC action beyond its core function in transcriptional control, and that it may participate in cytoplasmic regulatory mechanisms of gene expression via association with mRNA and protein complexes mediating PTR. Unquestionably, much remains to be tested to understand the molecular basis of this cytoplasmic, PTR-related function of GR and ultimately, its relevance in the global anti-inflammatory action of GC. On the structural level, it will be important to identify the GR domain interfacing with the GC-rich motif and the stoichiometry of this binding, as the GR lacks a conserved RNA-binding domain (RBD) that for other transcription factors/RBPs, like NF-90 and nucleolin, mediates association with mRNA (Ginisty et al. 1999; Shi et al. 2005). Furthermore, it will be necessary to define potential interactions with other RBPs and miRNAs that could mediate GR participation to RNPs, in addition to that mediated by RNA-bound mechanisms, or change its affinity for RNA binding. From a functional standpoint, it remains to be tested if acceleration of mRNA decay occurs for most of the putative GR motif-bearing targets identified, whether it is the only RNA-associated action of GR and whether other GR motifs, or RNP interactions, can mediate alternative cytoplasmic functions of the GR. It can be postulated that GR association with mRNAs can be modified by stimulus-induced changes in the composition of the RNP complex, and/or by action upon the association of GR-bound RNA with cytoplasmic foci of regulated mRNA decay and translation, such as P bodies and stress granules, where these functions are critically regulated during inflammation (Anderson and Kedersha 2009).
Related posts:
 Translational and Post-translational Control of Leptin Production by Fat Cells
Translational and Post-translational Control of Leptin Production by Fat Cells
 MicroRNA Regulation of Endocrine Functions in the Ovary
MicroRNA Regulation of Endocrine Functions in the Ovary
 Post-transcriptional and Post-translational Regulation of Steroidogenesis
Post-transcriptional and Post-translational Regulation of Steroidogenesis
 Post-transcriptional Regulation of Prostaglandin Biosynthesis
Post-transcriptional Regulation of Prostaglandin Biosynthesis
 Post-transcriptional Regulation of Insulin and Insulin Like Growth Factors
Post-transcriptional Regulation of Insulin and Insulin Like Growth Factors
 Mechanisms of Post-transcriptional Gene Regulation
Mechanisms of Post-transcriptional Gene Regulation
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


