Fig. 12.1
Overview of steroidogenesis. Tropic hormones bind to their respective G protein-coupled receptors leading to activation of the cAMP-PKA signaling cascade which in turn induces acute and/or chronicle effects on the regulation of steroidogenesis. Upon hormone stimulation, cholesterol acquired from endocytic or selective uptake pathways or from de novo synthesis is mobilized from lipid droplets and transported to the inner membrane of mitochondria through the collective actions of HSL, StAR and their associated proteins. At the IMM, cytochrome P450 side chain cleavage enzyme catalyzes the cleavage of a 6-carbon unit from cholesterol to produce pregnenolone, which is effluxed from mitochondria to the endoplasmic reticulum and used as the precursor for the synthesis of all of the other steroid hormones
The major synthetic pathways for steroid hormones in the adrenal gland and gonads are shown in Fig. 12.2. Although the final steroid product differs in a tissue-specific manner, the first committed reaction in the biosynthetic pathway is the same, i.e., the conversion of cholesterol to pregnenolone by the cytochrome P450scc enzyme (CYP11A1). P450scc is an enzyme complex consisting of a flavoprotein (NADH-adrenodoxin reductase), a ferredox (adrenodoxin) and a cytochrome P450 localized on an inner mitochondrial membrane (Miller 1988, 2008). P450scc catalyzes three distinct reactions: 20α-hydroxylation, 22-hydroxylation and scission of 20, 22 carbon–carbon bonds, thus converting cholesterol to pregnenolone (Miller 1988, 2008). Next, the pregnenolone is converted to the final hormone product by sequential steps along the pathway depending on the enzymes that are present in that tissue (Fig. 12.2). Thus, adrenal zona fasciculata-reticularis and zona glomerulosa cells produce cortisol (corticosterone in rodents) and aldosterone, respectively. Likewise, ovarian theca cells secrete androgens, while granulosa cells synthesize progesterone and estradiol. Testicular Leydig cells primarily synthesize and secrete testosterone.
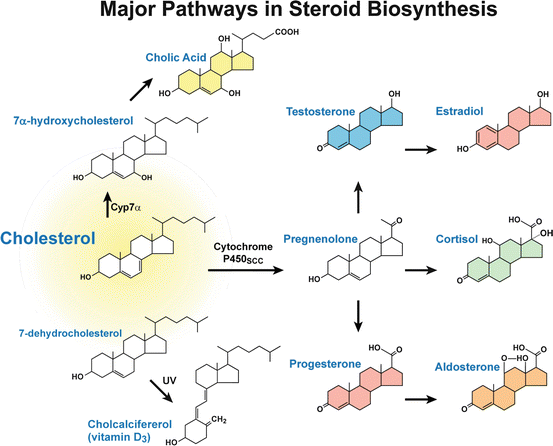
Fig. 12.2
The major synthetic pathways for steroid hormones
1.3 Post-transcriptional/Post-translational Regulation of Steroidogenesis
While much is known about the transcriptional regulation of steroidogenesis, relatively little is known about the post-transcriptional and post-translational regulation of this process. Below we summarize what is currently known about the post-transcriptional/post-translational modulation of steroidogenesis:
1.3.1 Phosphorylation/Dephosphorylation-Dependent Regulation of Steroidogenesis
Reversible phosphorylation of proteins, which can result in a conformational change in the structure in many enzymes and receptors, and in turn switches enzymes and receptors “on” and “off”, is an important regulatory mechanism that occurs in both prokaryotic and eukaryotic organisms (Krebs and Fischer 1964; Fischer and Krebs 1966; Burnett and Kennedy 1954; Bahler et al. 1990). Phosphorylation commonly occurs on serine, threonine, tyrosine and histidine residues in eukaryotic proteins (Ciesla et al. 2011; Hanks et al. 1988; Cohen 2000). Several key transcription factors and enzymes that contribute to the regulation of steroidogenesis, including steroidogenic transcription factors, SF-1, DAX-1, CREB and TORC (Babu et al. 2000; Desclozeaux et al. 2002; Sands and Palmer 2008; Takemori and Okamoto 2008), StAR (Arakane et al. 1997), HSL (Kraemer and Shen 2002) and downstream enzymes such as CYP11A1 and CYP17A1 (Miller 2008; Vilgrain et al. 1984), have been shown to be regulated via phosphorylation/dephosphorylation mechanisms.
(1a) Phosphorylation of StAR:
Orme-Johnson and colleagues first identified StAR in ACTH treated primary rat adrenocortical cell suspensions (Pon et al. 1986; Krueger and Orme-Johnson 1983; Epstein and Orme-Johnson 1991b). Using a two-dimensional gel electrophoresis of 35S-radiolabeled proteins, a hormone-induced phosphoprotein was identified by radioautography, termed ib and later as pp30. The synthesis of this protein was shown to occur with similar kinetics and dose response as corticosterone production after trophic hormone stimulation (Mellon and Griffin 2002; Stocco 2001; Manna et al. 2009). Subsequently, these seminal findings were further confirmed in the steroidogenic cells of the ovary and testis (Epstein and Orme-Johnson 1991a; Pon and Orme-Johnson 1998). Stocco and colleagues cloned this protein and renamed it steroidogenic acute regulatory protein, StAR (Clark et al. 1994). A number of in vitro studies have established that cAMP-dependent phosphorylation of murine StAR at serine residue 194 (Ser194; Ser195 in human StAR) is required for its maximal biological activity (Arakane et al. 1997; Fleury et al. 2004). Very recently, investigation into the functional importance of Ser194 phosphorylation in vivo using transgenic mice expressing either wild-type (WT) StAR or StAR mutation S194A to rescue StAR deficient (knockout) mice has been reported (Sasaki et al. 2014). The results showed that, despite protein expression comparable to or higher than amounts seen with control animals or rescued with WT StAR, mutant S194A StAR did not rescue the neonatal lethality and only partially rescued the sex reversal in male mice observed uniformly in StAR KO mice. Like the StAR KO mice, the adrenal cortex and testicular Leydig cells contained abundant lipid droplets when tissue sections were stained with Oil red O for lipids. Moreover, adrenal StAR from S194A rescued animals lacked an acidic species, which was detectable in response to ACTH treatment in animals rescued with WT StAR. These results are consistent with defective StAR phosphorylation. On the other hand, the WT-StAR transgene consistently restored viability and steroidogenic function in StAR-deficient (−/− mice) (Sasaki et al. 2008). These findings further establish that phosphorylation of Ser194 is essential for normal functioning of StAR protein in the adrenal cortex, testes and possibly ovaries of mice.
(1b) Phosphorylation of HSL:
Hormone sensitive lipase (HSL) was shown to be the neutral cholesterol esterase for the hormone-induced mobilization of stored CEs to supply precursor cholesterol for steroidogenesis (Kraemer and Shen 2002; Kraemer et al. 2002, 2004). Investigation into the functional significance of HSL in steroid production using HSL−/− mice showed that the cholesteryl ester content was substantially elevated in adrenals of HSL−/− mice, and basal corticosterone production was reduced approximately 50 %. The maximum corticosterone production induced by dibutyryl cAMP and lipoproteins was approximately 75–85 % lower in adrenal cells from HSL−/− mice compared with control. There is no intrinsic defect in the conversion of cholesterol into steroids in HSL−/− mice. Dibutyryl cAMP-stimulated conversion of high-density lipoprotein CEs into corticosterone was reduced 97 % in HSL−/− mice (Kraemer et al. 2004). The cholesteryl esterase activity of HSL is regulated by reversible phosphorylation/dephosphorylation (Yeaman 1990). A number of publications have reported PKA catalyzed phosphorylation of HSL at Ser563, Ser659 and Ser660 in rat HSL, resulting in increases in HSL hydrolytic activity (Yeaman 1990; Shen et al. 1998; Anthonsen et al. 1998). In addition, ERKs can phosphorylate adipocyte HSL on Ser600 and stimulate its activity (Greenberg et al. 2001). In contrast to activation of activity seen with PKA or ERK phosphorylation, other kinases, such as glycogen synthase kinase-4, Ca++/calmodulin-dependent protein kinase II, and AMP-activated protein kinase, phosphorylate HSL at a secondary basal site Ser565 in rat HSL (Yeaman 1990). Phosphorylation at Ser565 interferes with the phosphorylation of Ser563 by PKA (Yeaman 1990). HSL activity can be inactivated by protein phosphatases as well. The most active phosphatases against Ser563 are phosphatase 2A and 2C, while Ser565 is predominately dephosphorylated by phosphatase 2A (Wood et al. 1993).
(1c) Phosphorylation of Key Steroidogenic Transcription Factors and Enzymes:
SF-1, DAX-1, CREB and TORC have all been shown to be important transcription factors involved in steroidogenic pathways. SF1 can be phosphorylated by Erk2 at Ser203 in response to multiple components of the MAPK pathway and results in upregulated transcriptional activity (Babu et al. 2000; Desclozeaux et al. 2002). CREB can be phosphorylated by PKA as well as salt induced kinase in response to cellular stimuli and results in transcriptional activation of multiple targets involved in steroidogenesis (Sands and Palmer 2008; Takemori and Okamoto 2008). A few of the downstream enzymes, such as CYP11A1 and CYP17A1 (Miller 2008; Vilgrain et al. 1984), have been shown to be regulated via phosphorylation/dephosphorylation mechanisms.
1.3.2 Modulation of Steroidogenesis through Protein–Protein Interactions
In living organisms, cellular functions are often conveyed, at both cellular and systemic levels, by a large number of protein complexes through specific interactions between protein partners. Protein complex assembly can result in the formation of homo-oligomeric or hetero-oligomeric complexes. During steroidogenesis, many proteins, including SR-BI, StAR, HSL, are involved in formation of unique protein complexes for their functions.
(2a) SR-BI: Dimerization and interaction with PDZ domain containing proteins
In response to hormone stimulation, SR-BI, the HDL receptor responsible for selective CE uptake, changes to oligomeric form to facilitate CE uptake (for simplicity, we use the term dimerization here to include the multiple forms of the SR-BI protein; i.e., dimers and higher order oligomers). In one of the earliest direct demonstrations of protein–protein interactions involving SR-BI, it was shown to exist as homodimers in microvilli-enriched adrenal plasma membranes from 17α-ethinyl estradiol (17α-E2) primed rats (Azhar et al. 2002). Subsequent studies demonstrated that SR-BI exists in dimeric and high order oligomeric forms in all steroidogenic and non-steroidogenic cells and tissue which are active in ‘selective’ uptake of HDL-CEs (Reaven et al. 2001, 2006). Direct functional evidence for SR-BI dimerization came from the observation that SR-BI exists primarily in a monomeric form with some dimer formation in normal rat adrenal tissue. ACTH stimulation increased the dimerization of SR-BI in this tissue along with increased selective CE uptake, and dexamethasone-induced loss of ACTH led dramatically to the loss of SR-BI, SR-BI dimers and selective HDL-CE uptake. These results, coupled with striking architectural changes of the microvillar compartment at the adrenocortical cell surface, suggest that SR-BI dimers may, in a very basic way, be associated with SR-BI sites of action and function.
Additional functional studies further revealed a strong correlation between the levels of SR-BI dimers and increased selective HDL-CE uptake in cells and tissues. Moreover, co-immunoprecipitation studies of epitope-tagged SR-BIs (SR-BI-cMyc and SR-BI-V5) confirmed that SR-BI can exist as homodimers (Reaven et al. 2006). The use of cross-linking agents provided additional evidence that SR-BI forms dimers in native steroidogenic cell lines (endogenous), as well as in a heterologous insect cell expression system (Reaven et al. 2001). Also, analysis of cellular extracts from SR-BI transfected HEK-293 cells or ACTH-treated Y1-BS1 cells by size-exclusion chromatography and sucrose density centrifugation demonstrated that a significant portion of SR-BI exists in dimeric and oligomeric forms. Morphological analysis by immunoelectron microscopy provided independent confirmation of SR-BI homodimerization. More specifically, when double tagged-SR-BI proteins (SR-BI-cMyc and SR-BI-V5) were co-expressed in HEK-293 cells and the different proteins were subsequently immunostained and identified with two differently stained gold particles, there was mixing and clustering of gold particles suggesting (1) that the proteins travel to the same cell location, and (2) that many of the gold particles are in exceedingly close physical contact, i.e., within the distance accepted for protein dimers by fluorescent resonance energy transfer (FRET) technique. Similar results were obtained when Y1-BS1 mouse adrenocortical cells were transfected with V5 and/or cMyc tagged-SR-BI proteins. Interestingly, SR-BI transfected Y1-BS1 demonstrated major architectural changes along with the formation of double membranes in flower like arrangements. Gold-labeled secondary antibodies against V5 or cMyc antibody localized SR-BI to these sites, and revealed substantial dimer formation of this protein–shown by close contact between gold particles (Reaven et al. 2004, 2006).
Further investigations concentrated on the contribution of the cysteine residues in the extracellular domain (ECD) of SR-BI either independently or in cooperation with the C-terminal domain on SR-BI dimerization. SR-BI contains a total of eight cysteine (C) residues (C21, C251, C280, C321, C323, C334, C384, and C470) and six of them are located in the ECD. Mutagenesis studies showed that C280, C321, C323 and C334 residues in the ECD are necessary for preserving normal SR-BI (HDL) binding activity, selective CE uptake, and/or cell surface expression. Interestingly, mutation of any of these four cysteine residues to serine resulted in a robust induction of SR-BI dimer formation, but they were rendered non-functional because these residues are most likely essential for optimal HDL binding and, hence, selective CE uptake (Hu et al. 2011).
(2b) SR-BI interaction with accessory proteins
Emerging evidence now indicates that accessory proteins are also required for the proper cellular expression of SR-BI and SR-BI-mediated HDL-CE transport and other functions (Ikemoto et al. 2000; Silver 2002; Kocher et al. 2003; Komori et al. 2008; Robichaud et al. 2008; Yuhanna et al. 2008; Zhu et al. 2008; Fenske et al. 2009; Eyre et al. 2010; Kocher and Krieger 2009). For example, it has been shown that PDZK1/NHERF3 regulates hepatic SR-BI stability and steady state protein levels (Hu et al. 2013a). Interestingly, PDZK1/NHERF3 is neither expressed nor essential for SR-BI abundance or its cellular localization in steroidogenic cells of the adrenal gland, ovary and testis (Kocher et al. 2003). Recently, our laboratory made an important discovery that two other NHERF family members, NHERF1 and NHERF2, negatively regulate the expression and function of SR-BI in steroidogenic cells of adrenal and gonads, as well as the liver (Hu et al. 2013a). More specifically, we showed that NHERF1 and NHERF2 mRNA levels decrease in response to cAMP stimulation, whereas mRNA levels of SR-BI are upregulated. Co-immunoprecipitation, colocalization, bimolecular fluorescence complementation, and mutational analysis all indicated that NHERF1 and NHERF2 form complexes with SR-BI protein and, as a result, inhibit SR-BI-mediated selective CE transport and steroidogenesis. The PDZ1 or PDZ2 domain of NHERF1, the PDZ2 domain of NHERF2, or the MERM domains of NHERF1/2 and an intact COOH-terminal PDZ recognition motif (EAKL) in SR-BI is needed for the interaction. Both NHERF1 and NHERF2 also inhibit the de novo synthesis of SR-BI (Hu et al. 2013a). In contrast, no effect of NHERF4 was noted on selective HDL-CE uptake or steroidogenesis. Collectively, these data establish NHERF1 and NHERF2 as SR-BI protein binding partners that play a negative role in the regulation of SR-BI expression, selective CE transport, and steroidogenesis (Hu et al. 2013a).
(2c) StAR-TSPO interaction
StAR facilitates the rate limiting step in steroidogenesis, i.e., delivery of substrate cholesterol from the outer to the inner mitochondrial membrane where P450scc resides for the production of pregnenolone. Although the exact mechanism of action of StAR protein in mediating cholesterol transfer across the mitochondrial membranes is not known, limited evidence has previously suggested that StAR might work in concert with several other proteins including peripheral benzodiazepine receptor (PBR)/18-kDa transporter protein (TSPO), voltage-dependent anion channel 1 (VDAC1), phosphate carrier protein, cAMP-dependent protein kinase 1α (PKA-RIα) and TSPO-associated acyl-coenzyme A binding domain containing 3 (ABCD3) protein by forming a protein complex on the OMM (Stocco 2001; Rone et al. 2009; Miller 2007; Bose et al. 2008). However, two recent reports have provided evidence that PBR/TSPO is not involved in the steroidogenic process (Tu et al. 2014; Morohaku et al. 2014).
(2d) StAR-HSL interaction and HSL oligomerization
A significant amount of cholesterol transferred by StAR to mitochondria for steroidogenesis is hydrolyzed through HSL from CEs stored in steroidogenic cells. Using in vitro glutathione S-transferase pull-down experiments, we have demonstrated a direct interaction between HSL and StAR via the N-terminal as well as a central region of StAR (Shen et al. 2003). In addition, the 37-kDa StAR was co-immunoprecipitated with HSL from adrenals of animals treated with ACTH. Co-expression of HSL and StAR in Chinese hamster ovary cells resulted in higher cholesteryl ester hydrolytic activity of HSL. Transient overexpression of HSL in Y1 adrenocortical cells increased mitochondrial cholesterol content under conditions in which StAR was induced. It is proposed that the interaction of HSL with StAR in cytosol increases the hydrolytic activity of HSL and that together HSL and StAR facilitate cholesterol movement from lipid droplets to mitochondria for steroidogenesis.
In a separate study, using sucrose gradient centrifugation and in vivo and in vitro protein–protein interactions, we have demonstrated that HSL exists as a functional dimer composed of homologous subunits. Dimeric HSL displayed approximately 40-fold greater activity against cholesteryl ester substrate when compared with monomeric HSL without any differences in affinity for the substrate. Truncations of HSL identified the importance of the N-terminal 300 amino acids, as well as other regions, in participating in the oligomerization of HSL (Shen et al. 2000).
(2c) StAR-14-3-3γ interactions
Recently, the adaptor/scaffold protein 14-3-3γ was shown to interact with StAR by binding to Ser194 of StAR and thus retain StAR in the cytosol and delay maximum steroidogenesis in a pattern opposite to 14-3-3γ homodimerization (Aghazadeh et al. 2012, 2014; Liu et al. 2006; Papadopoulos et al. 2007). Under basal conditions, StAR resides in the cytosol and 14-3-3γ exists as homodimers; upon hormone stimulation, phosphorylation and acetylation of 14-3-3γ on residue Ser58 and Lys49 increase its binding with StAR, which delays maximum steroidogenesis. After 2 h, this interaction falls apart, allowing induction of StAR activity and steroid production to proceed at a maximal rate.
1.3.3 Modulation of Steroidogenesis by miRNAs
Recently, we and others have shown that SR-BI is post-transcriptionally regulated by microRNAs (Hu et al. 2012, 2013b). In addition, miRNAs have been identified that regulate LH/hCG receptor and steroidogenic enzymes in a tissue specific manner (Menon et al. 2013).
MicroRNAs (miRNAs ) comprise a novel class of endogenous non-protein-coding single-stranded small RNAs approximately 22–25 nucleotides long that have emerged as key post-transcriptional regulators of gene expression (Ambros 2004; Bartel 2004, 2009; Kim 2005; Fabian et al. 2010). They are transcribed in the nucleus by RNA polymerase II or RNA polymerase III into primary transcripts (pri-miRNAs) (Schwarz et al. 2003; Du and Zamore 2005, 2007), which are generally capped, polyadenylated and contain a hairpin stem of 33 bp, a terminal loop and two single-stranded flanking regions (Cai et al. 2004). These pri-miRNAs are then processed sequentially in the nucleus and cytoplasm by a complex of RNase III-endonucleases Drosha and Dicer to generate pre-miRNAs, and mature miRNAs, respectively (Siomi and Siomi 2010; Finnegan and Pasquinelli 2013). miRNAs cause post-transcriptional repression of protein synthesis by pairing with partially complementary seed sites in the 3′-untranslated regions (UTRs ) of target mRNAs, leading to either deadenylation and subsequent mRNA degradation and/or translational inhibition (Bartel 2004, 2009; Fabian et al. 2010; Bushati and Cohen 2007; Eulalio et al. 2008; Filipowicz et al. 2008; Ghildiyal and Zamore 2009). Importantly, a single miRNA can regulate expression of hundreds of target genes (Krishnan et al. 2013; Venkataraman et al. 2013), whereas the expression of a single gene can be regulated by multiple miRNAs (Hu et al. 2012; Gillen et al. 2011). Since the their original discovery in 1993, miRNAs has been shown to be key post-transcriptional regulators of gene expression in metazoan animals, plants, protozoa and viruses (Bushati and Cohen 2007). In mammals miRNAs are suggested to control the activity of more than 60 % of all protein coding genes (Friedman et al. 2009), and to participate in diverse cellular processes including development, cell-cycle control, metabolism, stem-cell differentiation, inflammation and immunity, oncogenesis, and diseases (Bushati and Cohen 2007; Barter 2009; Ambros 2004; Fabian et al. 2010; O’Connell et al. 2010; Yi and Fuchs 2011; Sayed and Abdellatif 2011; O’Connell et al. 2012; Abe and Bonini 2013; Di Leva et al. 2013; Fernández-Hernando et al. 2013; Flowers et al. 2013; Hata 2013; Szabo and Bala 2013; Rottiers and Naar 2012). Accumulating evidence now suggests that miRNAs also participate in the regulation of steroidogenesis (Menon et al. 2013; Yao et al. 2010; Romero et al. 2008; Robertson et al. 2013; Schmitz et al. 2011; Velazquez-Fernandez et al. 2014; Sirotkin et al. 2009; Yin et al. 2012; Yin et al. 2014; Dai et al. 2013; Xu et al. 2011; Kitahara et al. 2013).
Here we review the expression of miRNA in different steroidogenic tissues and their potential involvement in the regulation of steroid biosynthesis (Table 12.1).
Table 12.1
Summary of reported roles of miRNA during steroidogenesis
miRNA | Tissue | Process(es)/target(s) | Ref. |
|---|---|---|---|
(1) Adrenal | |||
miRNA-132, miRNA-212 | Adrenal | Aldosterone secretion | Hu et al. (2013b) |
miRNA-21 | Adrenal | Aldosterone secretion | Romero et al. (2008) |
Adrenocortical cells | |||
miR-24 | Adrenal | Cortisol and aldosterone production | Robertson et al. (2013) |
Adrenal cortical cells | CYP11B1 and CYP11B2 | ||
Human studies with pathophysiological implications | |||
miR-675, miR-139-3p | Adrenal | Discriminating ACCs from ACAs | Schmitz et al. (2011) |
miR-335 | |||
miR-21, miR-10b | Adrenal | Diagnose for ACC development, progression | |
miR-1395p, miR-let-7f | |||
(2) Ovary | |||
miR-21, miR-23a, miR-145, miR-503 | |||
miR-224, miR-383, miR-378, miR-132, and miR-212 | Equine follicle development | ||
miR-122, miR-136-3p | Ovary | LRBP, LHR | |
miR-455, miR-125a | Ovary | SR-BI | Hu et al. (2012) |
miR-133b | Ovary, granulosa cells | Foxl2, changes StAR and CYP19A level | Dai et al. (2013) |
miR-513a-3p | Ovary | LHCGR | Troppmann et al. (2014) |
miRNA-143 | Ovary, follicle development | ||
miRNA-145 | Ovary, granulosa cells | Activin receptor 1B, Smad 2 | |
miRNA-181a | Ovary, granulosa cells | Activin receptor IIA | Zhang et al. (2013) |
miRNA-224 | Ovary, granulosa cells | TGF-b, Smad4 | Yao et al. (2010) |
miRNA-320, MiRNA 383 | Ovary, granulosa cells | Targets E2F, SF1, suppress granulosa cell proliferation | |
miR-383 | Ovary, granulosa cells | RBMS1, c-Myc | |
Human studies with pathophysiological implications | |||
miR-132, miR-320, miR-miR-24, miR-222 | Ovary, follicular fluid | Regulates estradiol concentration | Sang et al. (2013) |
miR-24, miR-193b, miR-483-5p | Regulates progesterone concentration | ||
miR-132, miR-320 | Follicular fluid | Down-regulated in follicular fluid of polycystic ovary patients | |
miR-320, miR-383 | Follicular fluid | Up-regulated in POC | |
(3) Testis | |||
miR-132, miR-212 | Testicular Leydig cells, MLTC-1 | Hu et al. (2013b) | |
miR-34a,miR-181b, miR-122a | Mouse testis | In response to oxidative stress | Fatemi et al. (2014) |
Human studies with pathophysiological implications | |||
miR-367-3p, miR-371a-3p | Human serum | Plasma biomarker for TGCT | Syring et al. (2014) |
miR-372-3p, miR-373-3 | |||
2 MiRNA Regulation of LH/hCG and SR-BI Receptors
Mevalonate kinase (Mvk) has been identified as a novel luteinizing hormone receptor (LHR) mRNA binding protein (LRBP ), which binds to the coding region of LHR mRNA and causes translational suppression , leading to accelerated degradation of ovarian LHR mRNA in response to treatment with trophic hormones (Menon et al. 2013; Nair et al. 2002). Recently, it was demonstrated that miR-136-3p and miR-122 contribute to LH/hCG-induced down-regulation of ovarian LHR mRNA (Menon et al. 2013; Kitahara et al. 2013). In addition, miR-513a-3p was shown to target the LH/hCG receptor and control the level of LHCGR expression by an inversely regulated mechanism at the post-transcriptional level (Troppmann et al. 2014).
In a search for miRNAs regulating the expression of SR-BI, we recently provided evidence that miRNAs, such as miR-125a and miRA-455, target SR-BI mRNA, thus post-transcriptionally and negatively regulate SR-BI-mediated selective delivery of HDL-cholesterol in steroidogenic cells and consequently inhibit SR-BI-mediated and HDL-supported steroidogenesis (Hu et al. 2012). In addition, miR-185, miR-96, and miR-223 have been implicated in the negative regulation of hepatic SR-BI (Wang et al. 2013a).
3 miRNA Expression in Steroidogenic Cells of Adrenal Gland, Ovary and Testis
(3a) Adrenal
In an effort to examine the role of miRNAs in steroidogenesis, the expression profiling of miRNAs in rat adrenals in response to in vivo treatment of animals with hormones, ACTH, 17α-ethinyl estradiol (17α-E2) or dexamethasone (Hu et al. 2013b), was examined. Forty five out of the 72 expressed rat miRNAs showed significant changes in the adrenal in response to ACTH treatment. Among these, 27 mature or precursor miRNAs were up-regulated and 18 mature or precursor miRNAs were down-regulated in ACTH-exposed versus control adrenals. Among the various adrenal miRNAs whose expression was altered in response to treatment of rats with any of these three hormones, the most robust effect was on the expression of miR-132 and miR-212 (3–4 fold induction) in response to ACTH treatment (Hu et al. 2013b). The precursor for miRNA-212 was also up-regulated. Real-time PCR (qRT-PCR) confirmed that miRNA-212, miRNA-183, miRNA-182, miRNA-132 and miRNA-96 were up-regulated by ACTH, and miRNA-466b, miRNA-214, miRNA-503 and miRNA-27a were down-regulated by ACTH. MiR-132, together with miR-212, comprises the evolutionary conserved miR-132/212 family, encoded from the same intron of a small non-coding gene that is located on chromosome 11 in mice, chromosome 10 in rats and chromosome 17 in humans. Mature miRNA-132 and miRNA-212 share the same seed sequence (Wanet et al. 2012; Remenyi et al. 2013). In cardiac fibroblasts, miRNA-132/212 can modulate the angiotensin II signaling pathway (Eskildsen et al. 2014). In adrenal, angiotensin II is an important modulator of adrenal zona glomerulosa cell function, including aldosterone production and cell proliferation. The hormone-mediated increases in miR-132 and miR-212 levels suggest the possibility that they might mediate some of the trophic hormone regulation of steroidogenesis. Likewise, the level of miR-21 has also been shown to be specifically up-regulated by angiotensin II, and its expression levels were correlated with increased aldosterone secretion and proliferation in adrenocortical cells (Romero et al. 2008). In a screen using non-diseased human adrenal and aldosterone-producing adenoma samples, miR-24 was shown to be differentially expressed. Further analysis showed that miR-24 was able to modulate CYP11B1 and CYP11B2 expression, as well as cortisol and aldosterone production in human adrenal cortex (Robertson et al. 2013).
Related posts:
 Translational and Post-translational Control of Leptin Production by Fat Cells
Translational and Post-translational Control of Leptin Production by Fat Cells
 MicroRNA Regulation of Endocrine Functions in the Ovary
MicroRNA Regulation of Endocrine Functions in the Ovary
 Translational Control in the Germ Line
Translational Control in the Germ Line
 Post-transcriptional Regulation of Insulin and Insulin Like Growth Factors
Post-transcriptional Regulation of Insulin and Insulin Like Growth Factors
 Mechanisms of Post-transcriptional Gene Regulation
Mechanisms of Post-transcriptional Gene Regulation
 Post-transcriptional Regulation of Parathyroid Hormone Gene Expression in Health and Disease
Post-transcriptional Regulation of Parathyroid Hormone Gene Expression in Health and Disease
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


