Chapter Outline
MODIFICATION OF THE TETRAPYRROLE SIDE CHAINS
ENZYMATIC OXIDATION OF PROTOPORPHYRINOGEN IX TO PROTOPORPHYRIN IX AND INSERTION OF IRON
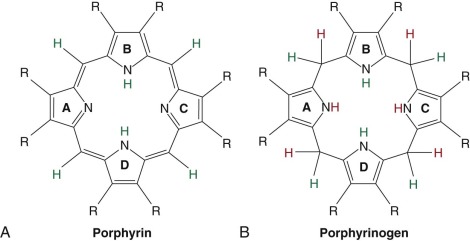
The porphyrias are caused by reduced activity of enzymes that constitute the heme biosynthetic pathway. With the exception of porphyria cutanea tarda (PCT), all are. Porphyrins are planar molecules comprised of four pyrrole rings connected by four methene groups. Porphyrins are also referred to as tetrapyrroles ( Fig. 12-1, A ). Porphyrins are intensely colored compounds (the word porphyria is derived from the Greek porphuros, meaning red-purple). All porphyrins avidly absorb light in a region of the spectrum known as the Soret band (around 400 nm) because of the alternating single-double bond structure of the porphyrin macrocycle. Once excited by light, porphyrins emit photons in the red region of the spectrum, a phenomenon responsible for their fluorescent properties. The emitted photons mediate the skin damage, which occurs in sun-exposed skin in some of the porphyric disorders.
The porphyrin precursors δ-aminolevulinic acid (ALA) and porphobilinogen (PBG) (a monopyrrole) are colorless compounds that do not fluoresce. Recent evidence suggests that these compounds are responsible for the neurovisceral manifestations of several of the porphyric disorders. A convenient classification scheme divides the porphyric disorders into three groups according to their clinical manifestations ( Box 12-1 ). Alternate schemes have been proposed by which the porphyrias are classified according to the organ in which the enzymatic defect is most apparent (hepatic porphyrias versus erythropoietic porphyrias), but these schemes are less useful to the clinician. Most heme synthesis takes place in erythroid precursors for the generation of hemoglobin, but approximately 15% of the daily production takes place in the liver, mainly for the formation of the cytochrome P450 enzymes. Heme is also a component of numerous other cellular proteins, including cytochromes, myoglobin, catalase, peroxidase, guanylate cyclase, and others, and the heme biosynthetic pathway is functional in all cells.
Photosensitivity Symptoms
Congenital erythropoietic porphyria (4) *
* Clinical manifestations usually recognized in childhood.
Porphyria cutanea tarda (5) †
† Clinical manifestations rarely recognized in childhood.
Hepatoerythropoietic porphyria (5) *
Erythropoietic protoporphyria (8) *
X-linked erythropoietic protoporphyria (1) *
Neurovisceral Symptoms
Aminolevulinate dehydratase porphyria (2) *
Acute intermittent porphyria (3) ‡
‡ Clinical manifestations rarely occur before puberty except in children with homozygous or compound heterozygous genotypes.
Photosensitivity and Neurovisceral Symptoms
Hereditary coproporphyria (6) ‡
Variegate porphyria (7) ‡
Numbers in parentheses designate the enzyme defect responsible for the disorder (see Fig. 12-2 )
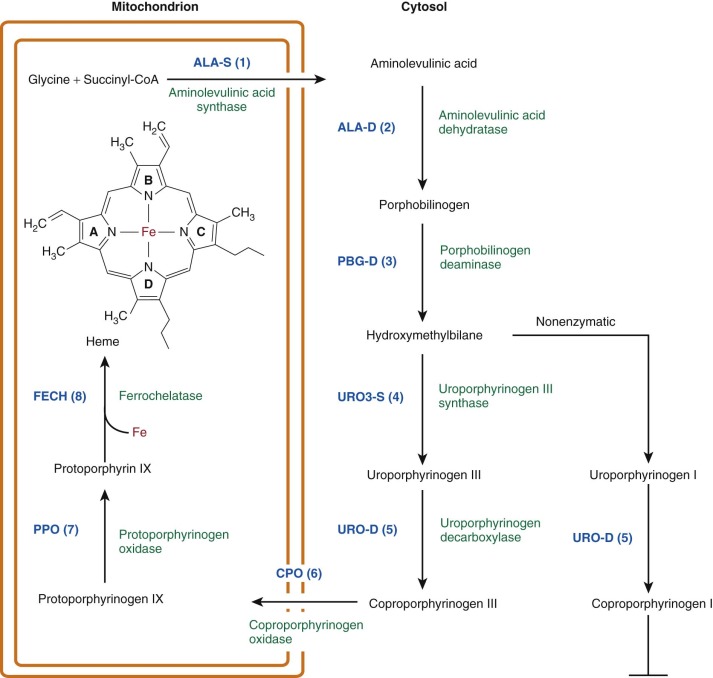
With the exception of ferrochelatase (FECH), the terminal enzyme in the pathway that catalyzes the insertion of iron into protoporphyrin IX (proto IX), all enzymes in the pathway that utilize cyclic tetrapyrroles require fully reduced porphyrinogens as substrates. Porphyrinogens are flexible, colorless, reduced compounds with hydrogen atoms added to each of the bridge carbons and to nitrogens of the pyrrole rings ( Fig. 12-1, B ). Porphyrinogens are very sensitive to oxidation. When the heme biosynthetic intermediates uroporphyrinogen and coproporphyrinogen are oxidized to the corresponding porphyrins, they can no longer serve as substrates ( Fig. 12-2 ) and are excreted in urine and stool. Protoporphyrinogen oxidase (PPO) also requires the reduced compound protoporphyrinogen as substrate.
Hans Fischer originally developed the currently utilized porphyrin nomenclature. Proto IX was the ninth isomer of protoporphyrin synthesized by Fischer and corresponded to the only isomer of protoporphyrin found in nature. Uroporphyrinogen and coproporphyrinogen could occur in four isomeric forms, but only the I and III forms occur in nature. The I and III isomers of uroporphyrinogen differ only in the positions of the acetate and propionate side chains on the D ring (see Fig. 12-1 ), but only the III isomer can ultimately be converted to proto IX and heme.
In eukaryotic cells, the heme biosynthetic pathway is divided between three compartments: (1) the mitochondrial matrix, (2) the mitochondrial intermembrane space, and (3) the cytosol (see Fig. 12-2 ). The pathway enzymes have been intensively characterized. All of the genes have been cloned, and many mutations responsible for the porphyrias have been identified. The crystal structures of all of the enzymes have been determined, providing models for the mechanisms of catalysis and the effects of specific mutations ( Table 12-1 ).
| Disease | Gene | Location | OMIM | E.C. # | PDB |
|---|---|---|---|---|---|
| None | ALA-S1 | 3p21.1 | 125290 | 2.3.1.37 | 2BWN * |
| X-Linked Sideroblastic Anemia, X-Linked EPP | ALA-S2 | Xp11.21 | 301300 | 2.3.1.37 | |
| ALA-D Deficiency Porphyria | ALA-D | 9q34 | 125270 | 4.2.1.24 | 1PV8 |
| Acute Intermittent Porphyria | PBG-D | 11q23.3 | 609806 | 4.3.1.8 | 3ECR |
| Congenital Erythropoietic Porphyria | URO3-S | 10q25.2 | 263700 | 4.2.1.75 | 1JR2 |
| Porphyria Cutanea Tarda, Familial Hepatoerythropoietic Porphyria | URO-D | 1p34 | 176100 | 4.1.1.37 | 1URO |
| Hereditary Coproporphyria | COP-OX | 3q12 | 121300 | 1.3.3.3 | 1R3Q |
| Variegate Porphyria | PROTO-OX | 1q22 | 600923 | 1.3.3.4 | 3NKS |
| EPP | FECH | 18q21.3 | 177000 | 4.99.1.1 | 2HRC |
Here we will dissect the complex heme biosynthetic pathway into four basic processes, and each porphyric disorder will be described in the context of the process that is defective:
- •
Formation of the pyrrole
- •
Assembly of the tetrapyrrole
- •
Modification of the tetrapyrrole side chains
- •
Enzymatic oxidation of protoporphyrinogen IX (proto’gen IX) to proto IX and insertion of iron
Formation of the Pyrrole
The δ-Aminolevulinate Synthases
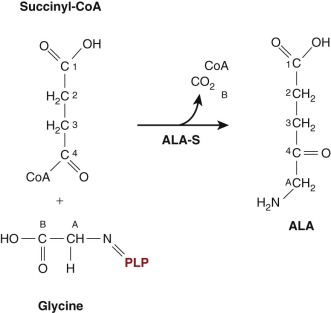
Regulatory mechanisms controlling heme synthesis in erythroid precursors and in the liver differ. In the liver, heme biosynthesis must respond rapidly to changing metabolic requirements. In erythroid cells, the pathway is regulated to permit a high steady state level of heme synthesis, and regulation is tied to the availability of iron. The first and rate-limiting enzyme in the pathway in all cells condenses glycine and succinyl-coenzyme A to yield ALA ( Fig. 12-3 ). This highly exergonic reaction occurs in the mitochondrial matrix and involves cleavage of the thioester bond of succinyl-coenzyme A. Two ALA-synthase (ALA-S) enzymes have evolved to accommodate the requirements of the liver (and other tissues) and the unique demands of the erythron. The first, ALA-S1, is expressed ubiquitously but at very low levels in erythroid precursors. The dominant form of ALA-S in erythroid cells is ALA-S2. The gene encoding ALA-S1 maps to the short arm of chromosome 3, whereas ALA-S2 is encoded on the X-chromosome. The two forms of ALA-S are 62% identical at the amino acid level. Although these two forms of ALA-S are regulated differently, both forms require pyridoxal-phosphate (PLP) as a cofactor. Pyridoxine deficiency is therefore associated with hypochromic anemia. Both ALA synthases function as homodimers.
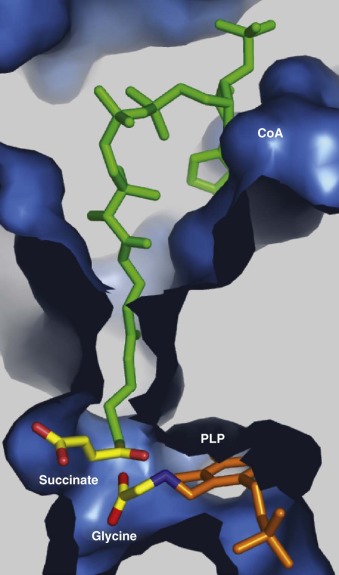
ALA-Ss are highly conserved between species, and the crystal structure of a bacterial ALA-S (from Rhodobacter capsulatus ) has been determined ( Fig. 12-4 ). The crystal structure supports the enzyme mechanism that had been suggested by biochemical studies.
In eukaryotes, both forms of ALA-S are synthesized in the cytosol and then translocated to the mitochondrial matrix, the site of succinyl-coenzyme A generation by the tricarboxylic acid cycle. Both forms contain an amino-terminal mitochondrial targeting sequence that also contains heme-binding domains designated as heme regulatory motifs (HRM). Binding of heme to the HRM inhibits mitochondrial import of both forms of ALA-S, representing a posttranslational point of end-product feedback inhibition of the pathway. The effect is greater for ALA-S1 than ALA-S2. In addition, developing erythroid cells express a heme transporter that prevents the accumulation of unbound heme in the cytosol (feline leukemia virus C receptor [FLVCR]).
Both ALA-S1 and ALA-S2 are transcriptionally regulated but by quite different mechanisms. Extensive studies in avian embryonic liver cells indicated that hemin, a chloride salt of heme, repressed transcription of ALA-S1, but the precise mechanism responsible for this effect has not been defined. Transcription of ALA-S1 is upregulated by the PGC-1α, a coactivator of nuclear receptors and transcription factors. Transcriptional regulation of ALA-S1 by PGC-1α is mediated by interaction of nuclear regulatory factor 1 (Nrf-1) and FOXO-1 (a forkhead family member) with the ALA-S1 promoter. When cellular glucose levels are low, transcription of PGC-1α is upregulated, leading to increased levels of ALA-S1 and creating conditions capable of precipitating acute attacks of the neurovisceral porphyrias. Upregulation of PGC-1α likely explains the clinical observation that fasting may precipitate attacks whereas glucose infusions are useful therapeutically.
There are two splice forms of ALA-S1 in humans. The major form lacks exon 1B ( Fig. 12-5 ) and is destabilized by heme. The minor form includes exon 1B and is resistant to heme-mediated destabilization. It is apparent that heme regulates ALA-S1 by multiple mechanisms, including transcription, translation, and translocation of the protein from the cytosol to the mitochondria. The regulatory effects of heme are mediated by the concentration of heme in small “pools” of heme not bound to hemoproteins. Factors depleting unbound heme pools result in induction of ALA-S1, an effect that is the prime factor in precipitating acute attacks of the neurovisceral porphyrias. No disease-causing mutations of ALA-S1 have been identified in humans, but down-regulating ALA-S1 with infusion of glucose and/or hemin forms the basis of therapy for acute porphyric attacks.
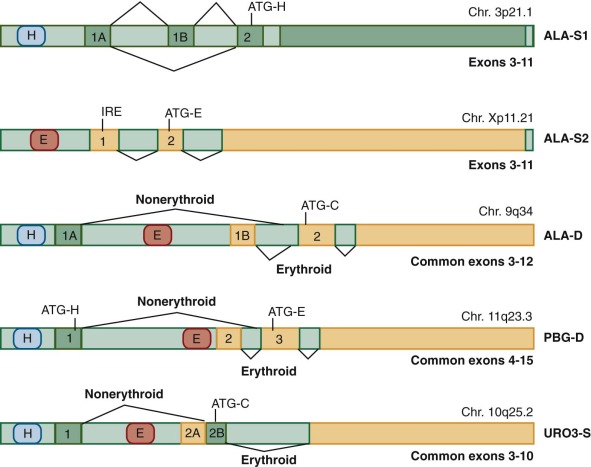
In contrast to ALA-S1, ALA-S2 is transcriptionally regulated by erythroid specific factors (including GATA1) that interact with promoter sequences found in many genes differentially regulated in red cells. Translational regulation also plays a major role in regulating expression of ALA-S2. The ALA-S2 transcript contains a 5′ iron responsive element (IRE), which interacts with an IRE-binding protein (IRP). The IRE-IRP complex prevents translation of the ALA-S2 mRNA. Addition of an iron-sulfur cluster (Fe/S) to IRP1 changes the conformation of the protein, reducing its affinity for the IRE. Fe/S clusters are generated and exported by mitochondria, linking regulation of heme biosynthesis in the red cell to the availability of iron and mitochondrial function. Loss-of-function mutations in ALA-S2 are causative for X-linked sideroblastic anemia (see Chapter 11 ). Gain-of-function mutations in ALA-S2 have been recognized as causative for X-linked erythropoietic protoporphyria (EPP) (see later discussion).
δ-Aminolevulinate Dehydratase
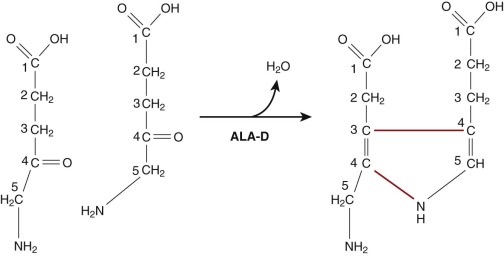
Once synthesized, ALA exits mitochondria by an unknown mechanism likely involving a transporter. In the cytosol, two molecules of ALA undergo a condensation reaction catalyzed by δ-aminolevulinate dehydratase (ALA-D) (also known as PBG synthase ) to form the monopyrrole PBG ( Fig. 12-6 ). The crystal structure of human ALA-D and ALA-Ds from many other species indicate that the enzyme functions as a tetramer of homodimers formed from eight identical subunits. Each of the homodimers contains one active site. Each of the eight subunits binds a zinc atom, and the enzyme is inactivated by replacement of the zinc by lead. Four zinc molecules are essential for catalysis. The remaining four zincs serve to stabilize the tertiary structure of the enzyme. Each active site binds two molecules of ALA through the formation of Schiff base linkages with active site lysine residues, one for each of the two substrate molecules. One molecule of ALA contributes the acetate side chain and the aminomethyl groups of PBG. The other contributes the propionate side chain and the pyrrole nitrogen (see Fig. 12-6 ).
In contrast to ALA-S, there is a single ALA-D gene located on chromosome 9q34 (see Table 12-1 ). The gene consists of two alternatively spliced noncoding exons (1A and 1B) and 11 coding exons (see Fig. 12-5 ). The translational start site is located in exon 2. A housekeeping promoter is located upstream of exon 1A, and a second erythroid-specific promoter is located between exons 1A and 1B. The erythroid promoter contains binding sites for erythroid specific transcription factors including GATA1 and Krüppel-like factor 1. Although the housekeeping and erythroid transcripts differ, they both encode identical proteins because they share the same translational start site in exon 2.
A common polymorphism of ALA-D, K59N, has been detected in approximately 10% of Caucasians, 3% of African Americans, and 6% to 11% of Asians. This polymorphism has been designated the ALA-D2 allele. The ALA-D2 allele retains normal enzyme activity and appears to have a higher affinity for both zinc and lead than the ALA-D1 allele. Lead-entering plasma is rapidly taken up by red cells and binds to ALA-D by displacing zinc and inactivating the enzyme. Multiple studies have shown an association between the ALA-D2 allele and high blood lead levels, but it remains unclear whether or not this allele is associated with increased susceptibility or increased resistance to lead toxicity.
Although the quaternary structure of human ALA-D is that of a homo-octamer, a homo-hexamer form of ALA-D composed of three homodimers has also been detected. The homo-hexamer has low enzyme activity. The octameric and hexameric forms of ALA-D are described as morpheeins, indicating that dissociation of the high activity octamer is followed by a conformational change and then reassembly as the structurally different low-activity hexamer. Conformational changes in the component dimers may result from substrate binding, but the most dramatic effects result from ALA-D mutations causative for ∂-aminolevulinate dehydratase deficiency porphyria (see later).
In all tissues, the enzymatic activity of ALA-D greatly exceeds the activity of ALA-S. The abundance of ALA-D in comparison with all of the heme biosynthetic enzymes has been ascribed to the finding that ALA-D plays several roles, serving as a proteasome inhibitor affecting the ATP/ubiquitin-dependent degradation of proteins, playing a role in chaperone assisted protein folding, and as a heme biosynthetic pathway enzyme.
δ-Aminolevulinate Dehydratase Deficiency Porphyria
ALA-D deficiency porphyria (ADP) is caused by homozygosity or compound heterozygosity for mutations of the ALA-D gene.
Frequency.
ADP is an autosomal recessive disorder resulting from loss of function mutations of ALA-D. The disorder is exceedingly rare, and fewer than 10 cases have been reported, two in one family. Most of the cases have represented compound heterozygotes. Eleven disease causing ALA-D mutations have been described, 10 of which are point mutations leading to amino acid substitutions. The sole exception is the deletion of two sequential bases resulting in a frame shift yielding a premature stop codon. Estimates of the frequency of heterozygosity for loss-of-function alleles of ALA-D, based on half-normal or less enzyme activity in erythrocytes of asymptomatic northern Europeans range from 1% to 2%. The extreme rarity of homozygosity or compound heterozygosity for ALA-D mutations suggests a high incidence of embryonic lethality when two mutant alleles of ALA-D are present.
Clinical Manifestations.
The clinical manifestations of ADP resemble those of the neurovisceral porphyrias but vary greatly in severity. The first two cases reported by Doss and colleagues in Germany were unrelated 15-year-old boys. Both suffered recurrent attacks of abdominal pain, nausea, vomiting, constipation, tachycardia, and a motor neuropathy causing paralysis. A third German case reported by Doss and colleagues also developed symptoms at age 15, including abdominal pain and a severe polyneuropathy. All three German patients were compound heterozygotes for ALA-D mutations.
The youngest reported case of ADP was a Swedish boy in whom muscle hypotonia was recognized at birth. At the age of 3, he developed recurrent attacks of abdominal pain, vomiting, and a motor polyneuropathy. This child also proved to be a compound heterozygote for ALA-D mutations. A liver transplant was performed at the age of 6 with the assumption that the metabolic defect was most profound in the liver. No change in the excretion of ALA was observed after the transplant, strongly suggesting that the red cells were the major source of this porphyrin precursor. The child died at the age of 9 of pneumonia and respiratory deficiency.
The oldest reported case of ADP was a Belgian man who developed a motor neuropathy and markedly increased urinary ALA excretion at the age of 63. ALA-D activity in erythrocytes was dramatically reduced to 1% of normal controls. The syndrome developed together with a clonal myeloproliferative disorder, namely polycythemia vera. Molecular studies revealed heterozygosity for a loss-of-function mutation (G133R) present in the polycythemia clone, which became clinically manifest with clonal expansion. This represents the only case in which a fully penetrant phenotype occurred in a heterozygote and supports the suggestion that the source of excess ALA in ALA-D porphyria is the erythron.
In two other cases, partial biochemical evidence of ALA-D deficiency were detected in heterozygotes but by a completely different mechanism. The first was a healthy Swedish newborn undergoing neonatal screening for hereditary tyrosinemia. Erythrocyte ALA-D activity was 12% of normal, and there were slight elevations of urinary ALA and coproporphyrin, as well as an increase in erythrocyte protoporphyrin. The child proved to be heterozygous for an F12L mutation, a catalytically inactive mutant that also affects secondary structure of the protein that favors formation of (with wild type protein) the low activity hexameric form of ALA-D. In spite of the ALA-D defect, the child remained healthy and developed normally.
The second case of partial biochemical expression in a heterozygote ALA-D mutant occurred in a 23-year-old American man and was also due to the F12L mutation. This patient presented with an acute porphyric attack clinically and biochemically compatible with hereditary coproporphyria (HCP). Indeed, a mutation of coproporphyrinogen oxidase (CPO) was identified. Because of a greater than usual increase in urinary ALA excretion and an increase in erythrocyte zinc protoporphyrin, an additional defect in ALA-D was suspected. Subnormal erythrocyte ALA-D activity was detected, and molecular studies revealed heterozygosity for the F12L mutation.
Pathogenesis.
The pathogenesis of the symptom complex associated with ADP is poorly understood. It has been suggested that ALA may act as a neurotoxin based on structural similarities between ALA and glutamate and ALA and γ-aminobutyric acid (GABA). High concentrations of ALA may interact with GABA receptors or inhibit glutamate uptake. ALA has been demonstrated to bind to GABA receptors in vitro, and GABA receptors are highly expressed by oligodendrocytes. In addition, oligodendrocytes are highly sensitive to glutamate-dependent metabolic pathways, which could be impaired by the effect of ALA on glutamate uptake. A possible role for ALA as a neurotoxin is supported in patients with hereditary tyrosinemia, who develop neurovisceral symptoms associated with marked inhibition of ALA-D by succinylacetone.
Laboratory Diagnosis.
Although ADP is rare, it should be suspected when an acute attack of abdominal pain suggests the possibility of a porphyric attack, especially when a prominent distal motor neuropathy is present. The dramatic reduction of ALA-D activity (to levels of 5% or less) results in the urinary excretion of large amounts of ALA and normal or minimally increased levels of PBG, a finding which distinguishes ADP from other neurovisceral porphyrias. Urinary excretion of coproporphyrin is markedly increased. The mechanism responsible for the increase in urinary coproporphyrin excretion is not known, but similar findings occur when ALA-D is inhibited in lead poisoning and when ALA is administered orally to normal individuals. Zinc-protoporphyrin accumulates in erythrocytes in patients with ADP, again by a poorly understood mechanism.
Heterozygous parents and siblings of probands with ADP have approximately half-normal ALA-D activity as measured in red cell lysates. Erythrocyte ALA-D assays are available in reference laboratories in the United States and Europe. Gene sequencing is available for ALA-D mutation analysis through the Department of Human Genetics at the Mount Sinai School of Medicine, New York, N.Y.
It is important to emphasize that ALA-D activity can be subnormal in disorders other than ADP. Patients with lead poisoning often develop clinical signs and symptoms similar to those found in patients with ADP and other neurovisceral porphyrias. Lead in plasma is taken up by red cells and inhibits ALA-D activity by displacing zinc atoms from the enzyme. The result is excess ALA in blood and urine, excess urinary excretion of coproporphyrin, and accumulation of zinc protoporphyrin in erythrocytes, findings also associated with ADP. The abnormal accumulation and excretion of porphyrins may be due, in part, to inhibitory effects of lead on CPO and FECH but is more likely due to disposition of the excess ALA, as occurs in ADP. Lead poisoning can be distinguished from ADP by measuring the blood lead level.
Patients with hepatorenal tyrosinemia type I, an autosomal recessive disorder due to deficiency of fumarylacetoacetate hydrolase (FAH), manifest acute episodes of porphyric-like neurologic crises and secrete large amounts of ALA in the urine. The FAH defect results in the generation of succinylacetone, a structural analog of ALA and a potent inhibitor of ALA-D.
A single patient with an acquired reversible, unidentified inhibitor of ALA-D, high urinary ALA levels, and a porphyria-like acute attack has been reported. In addition, both styrene and bromobenzene have been shown to inhibit ALA-D in rats. Humans exposed to these compounds have moderately decreased erythrocyte ALA-D activity but do not suffer clinical consequences.
ADP is a rare disorder and must be differentiated from other causes of ALA-D deficiency. Biochemical and molecular evidence is essential to establish the diagnosis of ADP.
Treatment.
Clear recommendations for the treatment of ADP are difficult because the number of reported cases is small, and the results of therapy have been variable. Suppression of ALA-S1 by the infusion of heme (heme arginate) proved effective in three German cases (all adolescent males), and infusions of glucose, narcotic analgesics, and hemin were effective in a 14-year-old American boy. Hemin infusion proved ineffective in the Swedish infant previously described. There have been no reports of marrow transplantation in a patient with ADP. Avoidance of drugs known to induce acute attacks in other neurovisceral porphyrias, generally by depleting hepatic heme pools and inducing ALA-S1, is suggested but has not been proven to be useful.
Assembly of the Tetrapyrrole
Porphobilinogen Deaminase
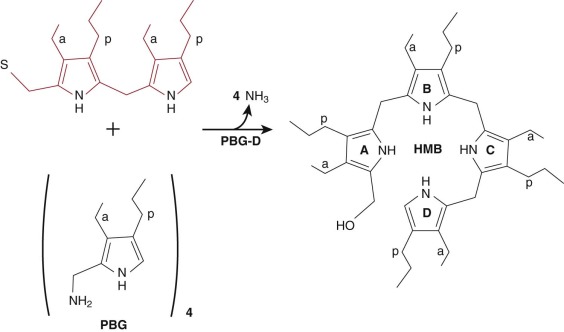
A polymer of four molecules of PBG is generated by PBG deaminase (PBG-D) (also called hydroxymethylbilane synthase ), a cytosolic enzyme. The polymer, an unstable tetrapyrrole, is designated hydroxymethylbilane (HMB) ( Fig. 12-7 ). PBG-D functions as a monomer, and the crystal structure of the human enzyme has been determined. Purification of PBG-D from both mammals and bacteria has yielded an enzyme with a dipyrrole (dipyrromethane) in the active site. The dipyrrole serves as a cofactor and is required for enzyme activity. A novel mechanism has been proposed to explain how PBG-D acquires the dipyrrole. PBG-D first binds HMB and then deaminates and polymerizes two additional molecules of PBG to form a hexapyrrole. The distal tetrapyrrole is cleaved and released as HMB. The proximal dipyrrole remains covalently bound through a thioester linkage and serves as the cofactor for subsequent rounds of catalysis. The source of the first HMB molecule to bind to PBG-D remains unexplained. Polymers of PBG can be formed nonenzymatically, but incubation of apo-PBG-D with PBG yields very little holoenzyme with the dipyrrole cofactor bound. It seems likely that the sequence of events involves nonenzymatic condensation of four molecules of PBG to form the amino-methylbilane, which interact with PBG-D. Although a poor substrate, amino-methylbilane is deaminated to form HMB, which is capable of forming a covalent thioester bond with PBG-D, leading to assembly of the initial hexapyrrole.
The human PBG-D gene has been cloned and mapped to chromosome 11q23-11qter. The gene spans 10 Kb and contains 15 exons. Two separate promoters control transcription and allow tissue specific regulation of gene expression, a mechanism also used by ALA-D and URO3-S (see Fig. 12-5 ). The housekeeping promoter lies upstream of exon 1 and is active in all tissues. The erythroid-specific promoter lies upstream of exon 2 and resembles other erythroid-specific promoters. Processing of the transcript initiated by the housekeeping promoter results in splicing of exon 1 to exon 3 and loss of exon 2. Transcription initiated by the erythroid promoter results in a transcript containing exons 2 through 15. Translation of the erythroid mRNA is initiated at an AUG located in exon 3, as exon 2 lacks an AUG codon. The AUG translational start codon in exon 1 of the housekeeping transcript is spliced in frame with the AUG in exon 3, resulting in a protein 17 amino acids longer at the amino terminus compared to the erythroid protein.
Acute Intermittent Porphyria
Acute intermittent porphyria (AIP) is caused by mutations of the PBG-D gene. Over 385 PBG-D mutations have been described. The spectrum of mutations includes point mutations, deletions, duplications, and insertions and has been found in the housekeeping promoter, exons 1 and 3 through 15, and in each of the 14 introns. Exon 12 appears to be a mutational hot spot with 46 identified mutations.
Frequency.
AIP is transmitted as an autosomal dominant trait, but five symptomatic children homozygous for disease–causing PBG-D mutations have been reported. Symptoms in these children began in infancy and included ataxia, dysarthria, psychomotor delay, and both central and peripheral neurologic manifestations. AIP is the most common of the acute neurovisceral porphyrias, but estimates of incidence vary according to the screening method used (clinical manifestations, erythrocyte PBG-D assays, molecular methods). Estimates based on a clinical diagnosis suggest an incidence of 0.13 per 1,000,000 in Europe. The incidence in Sweden is approximately four times higher, mainly due to a founder effect in northern Sweden. When subnormal erythrocyte PBG-D activity is used for screening, the incidence of AIP in European populations is much higher, but estimates based on erythrocyte PBG-D activity must be interpreted with caution. The normal range is wide, with considerable overlap between low-normal and high activity carrier values. Additionally, improper sample processing, storing, and shipping leads to decreased enzymatic activity. Molecular screening for PBG-D mutations on a large scale in a general population has not been done. It is likely that the incidence of undetected gene carriers in the population is much higher than the incidence of clinically evident AIP because most gene carriers remain symptom free throughout their lives. Recent estimates suggest a penetrance of only 1%. In all series, clinical symptoms occur more commonly in women than men.
Clinical Manifestations.
Clinical manifestations of AIP may occur before puberty in homozygotes or compound heterozygotes for PBG-D mutations, but most cases occur after puberty. Pediatricians are likely to encounter adolescent patients with symptomatic AIP, particularly adolescent girls following menarche. Virtually all of the clinical signs and symptoms that characterize attacks of AIP are due to neurovisceral dysfunction ( Table 12-2 ). Diffuse abdominal pain, often accompanied by nausea, vomiting, abdominal distention, and constipation, is the most common symptom of the acute attack. Abdominal pain is usually unremitting and severe but occasionally may be cramping. Decreased bowel sounds and even signs of ileus are generally present, but some attacks may be associated with enhanced gut motility and diarrhea. Tachycardia is often present. Other symptoms due to sympathetic over-activity or autonomic neuropathy include hypertension, tremors, sweating, and restlessness. Anxiety is a particularly common symptom, both during acute attacks and between attacks. A variety of other neuropsychiatric symptoms associated with AIP have been described, ranging from hypomania to overt psychotic behavior, and psychiatric symptoms may be the only manifestation of biochemically evident AIP.
| Signs and Symptoms | Frequency * | Comments |
|---|---|---|
| Gastrointestinal | ||
| Abdominal pain | Common | Generally constant and not localized |
| Nausea and vomiting | Common | Accompanies abdominal pain |
| Constipation | Common | Urinary retention may occur also. |
| Diarrhea | Uncommon | |
| Neurologic | ||
| Pain in back, chest, neck, head, extremities | Occasional | Pain may migrate to abdomen; may be accompanied by a sensory neuropathy |
| Paresis | Common | More proximal than distal; arms involved more often than legs |
| Respiratory paralysis | Uncommon | Occurs with progressive motor neuropathy and paresis |
| Mental symptoms | Occasional | Ranges from agitation to psychosis |
| Convulsions | Uncommon | Mainly occurs with hyponatremia |
| Cardiovascular | ||
| Tachycardia | Common | Rate control with β-blockers may be needed. |
| Systemic arterial hypertension | Occasional | May become chronic and affect renal function |
* Common: greater than 61%; Occasional: 21%-60%; Uncommon: less than 20%
A peripheral neuropathy may occur with or without an acute attack of abdominal pain. The neuropathy, due to axonal degeneration, is manifest mainly by muscle weakness in the extremities. Nerve conduction abnormalities may be minor, even with symptomatic weakness, but sophisticated studies of slow motor fiber conduction have been abnormal, even in the absence of symptoms. Cranial neuropathies are uncommon, but blindness due to optic nerve involvement or occipital lobe involvement has been reported.
Seizures may occur during an acute attack and are often associated with hyponatremia. When serum sodium levels are normal it is difficult to determine if the seizure is due to porphyria or to coexistent idiopathic epilepsy. In one large study of 268 patients, the lifetime prevalence of seizures was 5.1% in those who had suffered at least one acute attack and 2.2% in known carriers. Seizures occurring during latent periods are likely to be related to AIP. It is essential to establish a firm diagnosis of idiopathic epilepsy in AIP patients who experience seizures because many antiseizure medications may precipitate acute attacks (see later; Table 12-3 ). If seizures are associated with the hyponatremia of an acute attack, prolonged antiseizure therapy is not necessary.
Hyponatremia is a common finding during acute attacks. Hyponatremia maybe due to inappropriate secretion of antidiuretic hormone, but renal salt wasting has been implicated in patients with the lowest serum sodium vales. Serum sodium values below 120 nmol/L are generally found only in severe attacks.
The likelihood of full recovery after an acute attack has increased dramatically over the last 25 years. This is undoubtedly due to the introduction of effective therapy with hemin or heme arginate (see “Treatment” section). The risk of death, however, has not been eliminated. A review of 136 patients hospitalized with acute attacks at the University of Minnesota between 1940 and 1998 (i.e., both before and after the introduction of hemin therapy) found that 29 (21%) had died during an acute attack. In a study from Finland and Russia, 7 of 55 patients (13%) died as the result of an acute attack between 1966 and 2002. (Hematin therapy was first employed in 1971, but not widely used until 1978. ) In another study of 47 Finnish patients ascertained between 1967 and 1989, 6 deaths (12.7%) occurred as the result of an acute attack.
An unexplained finding in patients with AIP, variegate porphyria (VP), and HCP is a 35- to 70-fold increased risk of developing hepatocellular carcinoma. Most of these tumors occurred in individuals with a history of acute attacks. It has been suggested that DNA damage induced by ALA may be responsible for these observations. An unusual feature of the hepatocellular carcinomas arising in porphyric patients is the absence of associated liver cirrhosis. Chronic sustained hypertension is also prevalent in AIP and may be associated with renal impairment.
Pathogenesis.
Heterozygotes for PBG-D mutations have approximately half-normal enzyme activity in all tissues, which is usually sufficient for heme homeostasis. The enzymatic defect becomes manifest when demand for hepatic heme is increased, and ALA-S1 is induced. The result is increased production of ALA and PBG, creating a situation where the half-normal activity of PBG-D becomes rate limiting, and PBG (and ALA) accumulate in hepatocytes, circulate in plasma, and are excreted in the urine. Drugs are the most commonly recognized factors that increase the demand for hepatic heme, especially those metabolized by hemoprotein enzymes of the cytochrome P450 family. Barbiturates, anticonvulsants, and steroid hormones are well recognized precipitating factors in patients with AIP, but other factors not directly connected with P450 metabolism have also been incriminated, including fasting, dieting, and the stress of an acute illness. Most drugs taken by patients with AIP and other neurovisceral porphyrias have been classified as unsafe, potentially unsafe, probably safe, or safe. Isolated case reports have resulted in many drugs being classified as unsafe or potentially unsafe, resulting in a need for caution in drawing conclusions. Some drugs, however, have repeatedly been associated with acute porphyric attacks and must be avoided. A selection of these is shown in Table 12-3 .
| ACE inhibitors | Mephenytoin |
| Barbiturates | Metaclopramide |
| Carbamazepine | Primidone |
| Chlorpropamide | Progesterone |
| Danazol | Rifampin |
| Diphenylhydantoin | Succinimides |
| Ergots | Sulfasalazine |
| Estrogens | Sulfonamides |
| Glutethimide | Sulfonylureas |
| Griseofulvin | Valproic acid |
* A complete list of unsafe and safe drugs is available at two websites: (1) www.porphyriafoundation.com and (2) http://www.porphyria-europe.com .
The current prevailing view is that symptoms of the porphyric attack result from the toxic effects of ALA and/or PBG rather than from a deficiency of heme in neurons. The most striking evidence in support of this is the beneficial effect of allogeneic liver transplant in patients with repetitive and disabling acute attacks refractory to more conservative treatments. An allogeneic liver transplant rapidly normalizes urinary excretion of ALA and PBG and eliminates recurrent neurovisceral attacks. Furthermore, when explanted livers from AIP patients were transplanted into nonporphyric patients, the recipients promptly developed biochemical and clinical evidence of AIP. These impressive reports support the causative role of hepatic overproduction of ALA and PBG in acute neurovisceral attacks, but it is inappropriate to consider liver transplantation as a broadly applied treatment, especially with the efficiency of hemin or heme arginate infusions (see later).
Laboratory Diagnosis.
The signs and symptoms of an acute porphyric attack are quite nonspecific, and the correct diagnosis requires both consideration of a neurovisceral porphyria and a highly specific laboratory test. An acute porphyria is often not considered until fruitless, expensive imaging studies (and often unnecessary surgical exploration) are complete.
Increased urinary excretion of PBG is the key diagnostic finding in patients suffering an acute attack of AIP, HCP, or VP. Most methods used to quantify urinary PBG excretion rely on formation of a violet pigment when the sample is reacted with p-dimethyl aminobenzaldehyde (Ehrlich’s aldehyde). Typical reference ranges for PBG excretion are 0-18 µmol/day (0 to 4 mg/day). Values for ALA range from 0 to 60 µmol/day (0 to 8 mg/day). In contrast, urinary PBG excretion during an acute attack generally range from 220 to 880 µmol/day, and ALA excretion ranges from 100 to 450 µmol/day. Recently liquid chromatography-tandem mass spectroscopy methods have been developed that are rapid, specific, highly sensitive, and simultaneously measure ALA and PBG in urine and plasma.
A simplified method for measuring PBG at the bedside or in the emergency department employs a small anion exchange column packed in a kit that includes all of the required reagents and a color chart. The trace PBG kit (Thermo Trace/DMA, Arlington, TX) can detect PBG at concentrations greater than 6 mg/L, and results correlated well with standard quantitative assays. The kit does not measure ALA.
Urinary porphyrin excretion (mainly uroporphyrin) is increased during acute attacks of AIP, mainly from nonenzymatic polymerization of PBG, but this is a much less specific finding than increased excretion of PBG and ALA. Fecal porphyrin levels are generally normal, but plasma levels may be increased. In contrast, increased urinary porphyrin excretion during an acute attack of HCP and VP consists mainly of coproporphyrin, and stool porphyrin excretion is also increased. Markedly increased stool coproporphyrin and protoporphyrin are found in VP. Fluorescence scanning of diluted plasma at neutral pH readily distinguishes VP from the other neurovisceral porphyrias, even during asymptomatic periods.
A word of caution is required in interpreting urinary porphyrin values. Minimal elevations of urinary coproporphyrin may be found in patients with liver disease, marrow disorders, and a host of other illnesses. An incorrect diagnosis of porphyria is commonly made by over-interpreting minimal elevations of urinary coproporphyrin, leading to a delay in establishing the true cause of abdominal pain and other neurovisceral symptoms.
Assays of PBG-D activity in erythrocyte lysates have been widely employed to confirm the diagnosis of AIP and to detect asymptomatic family members once an index case has been identified. Erythrocyte PBG-D activity is approximately half-normal in most patients with PCT. Normal PBG-D activity, however, does not exclude a diagnosis of AIP because of differential splicing of the PBG-D transcript in the liver and the red cell. Mutations in exon 1 of the PBG-D gene affect activity of the enzyme in the liver, but the erythrocyte enzyme is encoded only by exons 2 through 15. It is important to emphasize that a valid half-normal measurement of erythrocyte PBG-D activity does not distinguish between asymptomatic (latent) and clinically manifest cases of AIP. Only urinary PBG levels can make this distinction.
PBG-D mutation detection by DNA analysis has proven to be more reliable than PBG-D enzyme assays and is now the preferred method for confirming a diagnosis of AIP and for identifying asymptomatic carriers. DNA studies have demonstrated that enzymatic assays may misclassify up to 15% of individuals tested. Mutation detection for AIP patients and their relatives is now readily available through the Department of Human Genetics at the Mount Sinai School of Medicine, New York, N.Y.
Treatment.
Treatment of an acute attack of AIP (and the other neurovisceral porphyrias) is designed to repress the activity of hepatic ALA-S1 and to offer supportive care and relief of symptoms. Intravenous infusions of heme result in a rapid reduction in levels of ALA and PBG in plasma and urine. Heme binds to plasma hemopexin and albumin and is then taken up by hepatocytes, where it expands the heme pool and represses ALA-S1. Many small, uncontrolled trials have demonstrated both biochemical and clinical responses to infusion of heme. In one study involving 22 patients suffering 57 acute attacks, all patients responded favorably, but therapy was most beneficial when administered early in the attack, a finding confirmed by others. A single, placebo-controlled trial found dramatic biochemical benefits of heme infusion, but clinical benefits were more difficult to confirm.
Heme is not soluble at neutral pH, but solubilized hemin (heme-chloride) and heme arginate have been administered to many patients. In the United States a lyophilized preparation of hemin, (Panhematin, Recordati Rare Diseases, Lebanon, NJ) has been approved by the U.S. Food and Drug Administration for the treatment of acute porphyric attacks. Hemin is unstable when reconstituted with sterile water, and degradation may cause an anticoagulant effect and phlebitis at the site of infusion. Phlebitis can be avoided by the use of intravenous catheters that extend to larger veins (e.g., the superior vena cava). Instability and the locally irritating effects of lyophilized hemin can be attenuated by solubilizing hemin in 25% albumin, a method now recommended. The recommended regimen for heme therapy is 3 to 4 mg/kg infused once daily on 4 consecutive days. Clinical improvement generally occurs within 1 or 2 days, but when neuronal damage is severe, complete recovery may take months. The keys to success are to initiate therapy as soon as an attack is recognized and to complete a 4-day course. Rare side effects of therapy include constitutional symptoms such as fever, myalgias, and malaise, but one case of marked hypotension and another of transient renal failure have been reported. The latter case was associated with an excessive dose (1000 mg) of hemin.
Heme arginate (Normosang, Orphan Pharmaceuticals, Paris, France) is more stable in solution than hemin, is equally effective, and is widely available in Europe and South Africa but remains an experimental drug in the United States. Phlebitis is a rare complication with heme arginate, and the shelf life is considerably longer than that of hemin. Accidental overdosing with heme arginate has been associated with acute hepatic failure. Adenoviral-mediated hepatic expression of PBG-D corrects the metabolic defect in mouse models of AIP, but no studies have been done in humans. Intravenous infusions of recombinant human PBG-D have been administered to normal subjects and asymptomatic PBG-D mutation carriers. Transient decreases in plasma concentrations of PBG were noted, and no serious side effects occurred. No trials of recombinant PBG-D for the treatment of symptomatic AIP have been reported.
Intravenous infusions of glucose (300 to 500 g/day, administered as 10% glucose) may be effective when an attack is mild or until hemin is obtained, but glucose is far less effective than hemin. Infusions of glucose in water solutions are contraindicated because they aggravate hyponatremia, which can prove fatal. The mechanism of the glucose effect relates to the previously described finding that transcription of ALA-S1 is upregulated by the peroxisome PGC-1α and transcription of PGC-1α is controlled by glucose availability. Insulin may downregulate PGC-1α, and it has been suggested that the combination of glucose and insulin may be more effective than glucose alone.
Acute attacks require treatment of symptoms in addition to therapy directed at ALA-S1. Pain is often severe, and full doses of morphine or other opioids are both safe and effective. Phenothiazines in modest doses are safe and are useful for the control of nausea and anxiety. Hypertension and tachycardia may respond to β-adrenergic blocking agents, but these agents should be used with caution when patients are hypovolemic because increased catecholamine secretion is often an important compensatory mechanism. Seizures during an acute attack can be a particularly vexing problem because most antiseizure drugs have the potential to worsen the intensity of the attack. Clonazepam and gabapentin may be safe, and benzodiazepines are probably safe. Hyponatremia and hypomagnesemia should be corrected because these abnormalities are common and may be causative for seizures.
The long-term management of patients with acute neurovisceral porphyrias is focused on the prevention of future attacks. Avoidance of unsafe drugs (see Table 12-3 ) is critical as is maintenance of a balanced, calorically adequate diet. In some women acute attacks occur only in the luteal phase of the menstrual cycle. In these cases gonadotropin-releasing hormone (GnRH) analogues can be highly effective. Prolonged use of GnRH analogues in young women may cause irreversible bone loss, but low-dose transdermal estradiol may safely prevent this complication. Both cyclic and noncyclic attacks may be prevented by frequent infusions of hemin, but the expense and the potential for iron overload (8 mg of iron per 100 mg of hemin) makes this an unattractive option.
Levels of progesterone, an inducer of hepatic heme synthesis, are elevated in pregnancy, but pregnancy is well tolerated by most women with acute porphyrias. In a large series reported from Finland of 176 pregnancies in women with AIP or VP, porphyric symptoms occurred in fewer than 10%. Worsening symptoms during pregnancy may be due to harmful drugs, inadequate nutrition, or both.
Uroporphyrinogen III Synthase
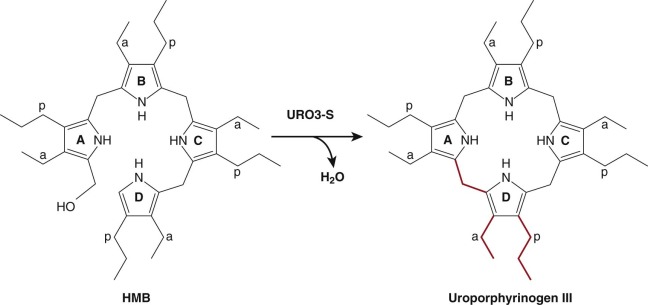
The product of the PBG-D reaction, HMB, serves as the substrate for uroporphyrinogen III synthase (URO3-S). URO3-S catalyzes the asymmetrical cyclization of HMB to the III isomer of uroporphyrinogen (uro’gen III). The reaction involves “flipping” of the D ring to yield the asymmetric product ( Fig. 12-8 ). Rearrangement of the D ring occurs by a complex mechanism involving cleavage and formation of carbon–carbon bonds. Spontaneous cyclization of HMB can also occur, but the product, uroporphyrinogen I (uro’gen I), cannot ultimately be converted to heme. URO3-S functions as a monomer. Although the primary sequence is highly divergent, the crystal structures of human URO3-S and a bacterial URO3-S are nearly identical. The structure of URO3-S from the bacterium Thermus thermophilus has been determined with the uro’gen III product bound. The structure suggests that the enzyme prevents formation of uro’gen I by positioning the A and D rings to favor formation of uro’gen III.
The human URO3-S gene is localized to chromosome 10q25.2-26.3 (see Table 12-1 ) and contains 10 coding exons. The translational start site is located in exon 2B (see Fig. 12-5 ). Both housekeeping and erythroid-specific transcripts are produced. The housekeeping promoter lies upstream of exon 1. The housekeeping transcript results from a splicing event that joins exon 1 and 2B. The erythroid-specific promoter lies within intron 1 and contains eight GATA1 binding sites. The erythroid-specific transcript contains all of exon 2. Although the housekeeping and erythroid transcripts are expressed from different promoters, they produce identical proteins.
Congenital Erythropoietic Porphyria
Congenital erythropoietic porphyria (CEP) (Günther disease) is a rare form of porphyria in humans due to mutations of URO3-S. About 200 cases have been reported. The disorder is of historical interest as it was the first porphyria to be recognized as an inherited trait and the first to be characterized biochemically. CEP also occurs in squirrels, cows, and other species. Over 40 URO3-S mutations have been reported, including exonic and promoter point mutations, deletions, insertions, and splice-site mutations.
Frequency.
The rarity of CEP makes frequency estimates difficult, but the disorder has been described in multiple ethnic groups. The frequency of heterozygosity for URO3-S mutations is not known because heterozygotes have no phenotype, and population-based sequencing screens have not been done. Although rare, CEP is recognized more frequently than ALA-D porphyria, perhaps because of the dramatic cutaneous phenotype.
Clinical Manifestations.
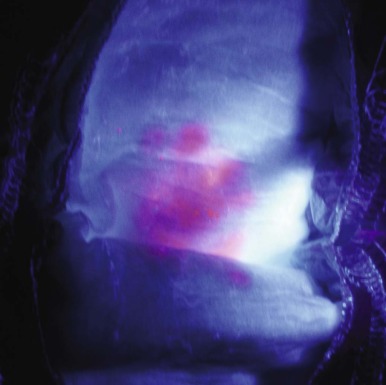
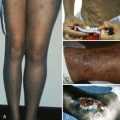
Cutaneous photosensitivity is the most dramatic feature of CEP and is usually recognized in early infancy. The diagnosis may be suspected because of the dark brown color of urine soaked diapers that demonstrate a distinctive, intense red-pink fluorescence when illuminated with long-range ultraviolet light (e.g., a Wood’s light) ( Fig. 12-9 ). The severity of the skin lesions varies widely and is related to the amount of residual URO3-S activity, which in turn determines the magnitude of porphyrin accumulation and excretion. In most cases photosensitivity is severe and is characterized by skin fragility, bullous lesions, ulcerations, and secondary infections. If exposure to sunlight is not rigorously avoided, mutilation of the face and hands is the ultimate outcome ( Fig. 12-10 ). A common finding in CEP and the other porphyric disorders associated with photosensitivity is the presumably protective response of extensive hypertrichosis. Photoinduced damage of nails and ocular structures may also occur. A unique finding in children with CEP is red-brown discoloration of the teeth, due to deposition of porphyrins into dentine and enamel during tooth development. The teeth fluoresce when illuminated with a Wood’s light.
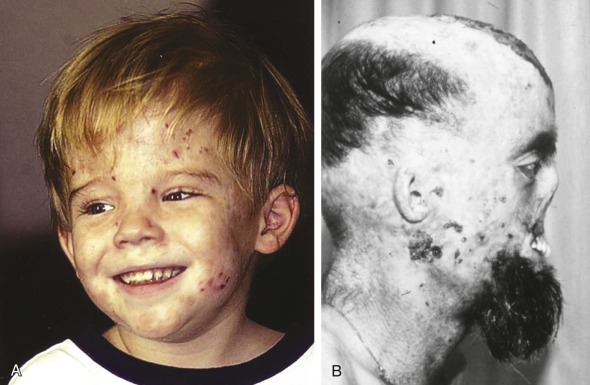
Hemolytic anemia of varying severity is found in nearly all cases, but detailed morphologic findings in the blood and bone marrow have been reported infrequently. Ineffective erythropoiesis may also be present. Poikilocytosis, nucleated red cells, and basophilic stippling are common, and the marrow demonstrates erythroid hyperplasia and may manifest marked dyserythropoiesis. Fluorescence microscopy may demonstrate fluorocytes in the peripheral blood, but this is more dramatic in the bone marrow. Splenomegaly generally accompanies the hemolytic anemia, and the magnitude of splenic enlargement reflects the severity of hemolysis. Transfusion dependence may develop. Leukopenia and symptomatic thrombocytopenia may occur as a result of splenic sequestration.
A variety of osteolytic skeletal lesions have been described when the disease reaches the stage of mutilation. Vitamin D deficiency as a result of sun avoidance may play a role in the development of osteolysis. At postmortem examinations the skeleton is fluorescent under long-range ultraviolet light.
Pathogenesis.
Loss-of-function mutations of URO3-S lead to the accumulation of the HMB substrate in erythrocyte precursors, which is readily converted nonenzymatically to uro’gen I. Residual activity of URO3-S is generally less than 10% of normal. Uro’gen I may be auto-oxidized to uroporphyrin I or may serve as a substrate for uroporphyrinogen decarboxylase (URO-D) ( Fig. 12-2 ), ultimately generating coproporphyrinogen I (copro’gen I). This results in a metabolic “dead end” as the next enzyme in the pathway, CPO, is stereospecific and will only accept coproporphyrinogen III (copro’gen III) as a substrate. Uroporphyrin I and to a lesser extent coproporphyrin I accumulate in erythroid precursors. Erythrocyte fragility and hemolysis result by an undefined mechanism, liberating porphyrins into the plasma. Uroporphyrin I mediates the cutaneous photosensitivity and is excreted in the urine. Coproporphyrin I is excreted in the stool and to a lesser extent in the urine.
Two phenotypic copies of CEP have been reported. In one, a partial defect in URO-D combined with a congenital dyserythropoietic anemia produced mutilating photosensitivity and massive uroporphyrinuria. The pattern of urinary porphyrin excretion differed from CEP as large amounts of heptacarboxyl porphyrin were found in addition to even larger amounts of uroporphyrin. In the other, a mutation in the erythroid specific transcription factor GATA1 resulted in subnormal URO3-S in red cells and a phenotype of CEP, thalassemia intermedia, and thrombocytopenia.
The clinical manifestations of CEP vary greatly, but a unique case has been reported in which a gain-of-function mutation of ALA-S2 coexisted with compound heterozygosity for loss-of-function mutations of URO3-S. The result was a severe CEP phenotype compared to other family members with the same URO3-S genotype.
Late Onset CEP.
CEP may become evident in adult life, and clinical symptoms are generally less severe than in the more typical early-onset cases. In most of the adult-onset cases, moderate URO3-S defects became evident only when an acquired clonal marrow disorder developed.
Laboratory Diagnosis.
Urinary porphyrin excretion is massively increased in CEP, in some cases up to 1000 times normal. The pattern of porphyrin excretion, determined by high-performance liquid chromatography is characteristic. The dominant urine porphyrin is uroporphyrin I with smaller amounts of coproporphyrin I. Assays of URO3-S have been performed in erythrocytes and cultured cells, but the assays are not widely available. The most practical way to confirm the diagnosis of CEP is through direct sequencing of the gene, a procedure available through the Department of Human Genetics at the Mount Sinai School of Medicine in New York, N.Y. URO3-S mutations can be detected in chorionic villi or cultured amniotic cells. This may be useful in subsequent pregnancies when parents have had a child with CEP.
Treatment.
The only curative therapy for CEP is an allogeneic marrow transplant. Sixteen patients with CEP who have undergone allogeneic transplants have been reported. The published experience suggests that early transplant is preferred, particularly when hematologic manifestations are prominent, but this approach is not universally applicable because of potential donor availability, treatment-related morbidity, and financial constraints. Allogeneic transplants should probably be restricted to the more severe cases of CEP because conservative management can yield longevity of 40 to 60 years.
The morbidity associated with marrow allografts could be avoided by autologous hematopoietic stem cell transplant if stem cells could be stably transfected with a normal URO3-S cDNA. This has been accomplished in a murine model of CEP using a lentivirus vector and in isolated keratinocytes from a CEP-affected individual. In the latter case, the lentiviral vector was excised, yielding induced pluripotent stem cells free of viral reprogramming genes. Complete correction of the phenotype was achieved in both the murine model and in the human pluripotent stem cells. No clinical trials in humans have yet been reported.
Avoidance of sunlight is at the core of conservative management. This can be achieved with protective clothing and effective sunscreens. Conventional sunscreens containing para-aminobenzoic acid are ineffective and opaque “pancake” makeup is preferred. Splenectomy may reduce the severity of hemolysis and correct leukopenia and thrombocytopenia, but beneficial effects on photosensitivity are often transient. Erythropoiesis and endogenous porphyrin production can be suppressed with aggressive transfusion regimens, but transfusion-induced iron overload results.
Modification of the Tetrapyrrole Sidechains
Uroporphyrinogen Decarboxylase
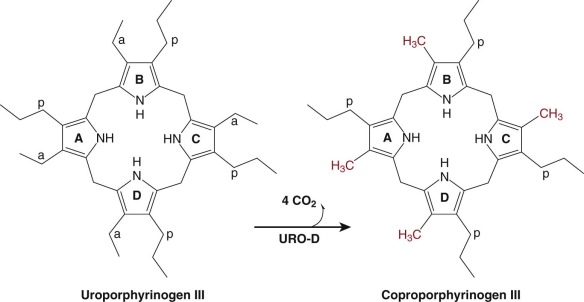
Uro’gen III is converted to copro’gen III by the sequential removal of the carboxyl groups of the four acetate side chains yielding four methyl groups. This reaction is catalyzed by the cytosolic enzyme URO-D ( Fig. 12-11 ). Decarboxylation of uro’gen III begins at the asymmetric D ring and proceeds in a clockwise manner. Uro’gen III is the preferred substrate. The enzyme will accept uro’gen I as a substrate, but the product, copro’gen I, cannot be converted to heme.
Human URO-D is encoded by a single gene located on chromosome 1p34 (see Table 12-1 ). Unlike ALA-D, PBG-D, and URO3-S, there is no erythroid specific promoter in the URO-D gene. The URO-D mRNA is highly expressed in developing erythrocytes, but the mechanism responsible for erythroid-specific upregulation is not known.
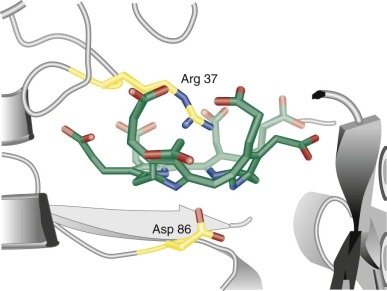
URO-D has been purified from human erythrocytes, bovine liver, and from yeast and bacteria. Unlike most decarboxylases, URO-D does not require cofactors or prosthetic groups for activity. The crystal structure of human URO-D has been determined, revealing that URO-D functions as a homodimer. Each monomer contains an active site cleft. The active site can only accommodate the flexible porphyrinogen macrocycle (see Fig. 12-12 ). Oxidation of the bridge carbons connecting the pyrrole rings renders the molecule rigid and planar and may explain why uroporphyrin is not a substrate for URO-D.
Porphyria Cutanea Tarda
PCT is due to subnormal activity of URO-D in the liver. The disorder is characterized clinically by a photosensitive dermatosis that is usually less severe than in CEP. Expression of the clinical phenotype is associated with one or more risk factors: iron overload, often due to homozygosity for the C282Y mutation of the hemochromatosis gene (HFE); hepatitis C; alcohol abuse; and the use of medicinal estrogens. The frequency of these risk factors is far higher than observed incidence of PCT, suggesting that susceptibility factors, either genetic or environmental, yet to be identified are important determinants of disease expression.
Two major variants have been identified. The first, designated familial PCT (F-PCT), is associated with heterozygosity for mutations at the URO-D locus. F-PCT is transmitted as an autosomal dominant trait. Affected individuals display approximately half-normal URO-D activity and half-normal URO-D protein in all tissues. There is considerable variation in URO-D activity because different mutations have varied effects on enzyme activity. F-PCT accounts for approximately 25% of the clinically observed cases. Most carriers of a mutant URO-D allele do not express a porphyric phenotype unless one or more of the risk factors mentioned above are also present. Clinical expression of F-PCT in childhood is rare. The authors have evaluated three children with F-PCT, two of whom were brothers, who were also homozygous for the C282Y HFE mutation.
The second major variant, and the most prevalent form of PCT, is designated sporadic PCT (S-PCT) and accounts for approximately 70% of cases. In S-PCT, reduced activity of URO-D is restricted to the liver. No URO-D mutations have been identified in patients with S-PCT, and there is no evidence of a liver-specific URO-D transcript. Nevertheless, S-PCT may cluster in families suggesting the presence of predisposing genetic factors. The concentration of URO-D protein is normal in all cells, including the hepatocytes, yet when the disease phenotype is expressed, enzyme activity is markedly reduced in the liver. Normal URO-D protein concentration coupled with subnormal URO-D enzymatic activity strongly suggests the presence of an enzyme inhibitor. Risk factors identified when S-PCT is clinically evident are identical to those associated with F-PCT.
A third rare variant of PCT occurs in children who have inherited two mutant alleles of URO-D. This variant has been designated hepatoerythropoietic porphyria (HEP) because porphyrins accumulate in both the liver and the red cells. Severely reduced levels of URO-D resulting from the genetic defect become rate limiting. Activity in red cell lysates is generally less than 10% of normal but may be as high as 20% in cases with a mild phenotype.
A fourth variant of PCT, designated toxic PCT, has been associated with exposure to halogenated aromatic hydrocarbons. An epidemic of toxic PCT occurred in Turkey between 1956 and 1961 following ingestion of wheat treated with the fungicide hexachlorobenzene. Many of the Turkish cases were children. Later studies in animals established that the halogenated aromatic hydrocarbons diminished hepatic URO-D activity. PCT in humans has also been reported following exposure to 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) and dichlorophenols and trichlorophenols.
Frequency.
PCT is the most common of the porphyric disorders, but the incidence varies in different populations. In Caucasian populations of northern European origin, the incidence may be as high as 2 to 5 per 20,000, likely reflecting the high incidence of hemochromatosis gene mutations in this group. In contrast, PCT is uncommon in African Americans but does occur in the Bantus of South Africa, who develop iron overload from beer brewed in iron pots. PCT occurred commonly in patients undergoing chronic hemodialysis, but substitution of erythropoietin for red cell transfusions (hence avoiding transfusion-induced iron overload) has reduced the incidence.
The high frequency of PCT, with one quarter of cases associated with URO-D mutations, predicts that HEP should not be rare. Instead fewer than 40 cases have been reported. This is best explained by embryonic lethality of extremely low residual activity of mutant URO-D enzymes or the lethal effect of null alleles.
Clinical Manifestations.
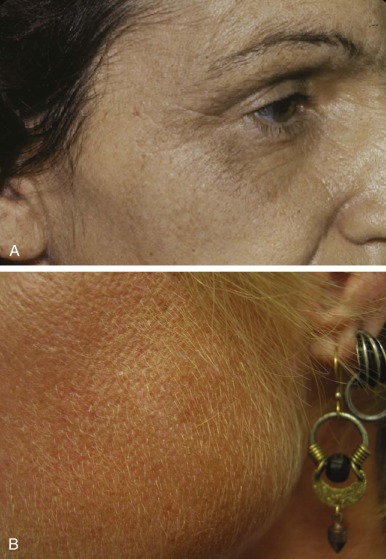
Skin fragility on the dorsal aspects of the hands is often the initial finding, but blistering lesions (bullae) then appear on all sun-exposed areas. The fluid-filled bullae rupture, and the denuded area heals slowly, often forming thick eschars and eventually hyperpigmented patches ( Fig. 12-13 ). Facial hypertrichosis is common, particularly in children and women. Facial hypertrichosis may be localized to periorbital regions ( Fig. 12-14 A ) or can be manifest in a lanugo-like diffuse facial pattern with or without pigmentation ( Fig. 12-14 B ). Some patients develop scleroderma-like skin changes, mainly in light-exposed areas. When PCT becomes chronic, milia (small, cystlike collections of keratin) are common on the hands. Similar photocutaneous findings have been seen in patients with pseudoporphyria, a disorder in which plasma and urine porphyrins are normal. The skin findings are likely due to other photosensitizing compounds. Nonsteroidal antiinflammatory drugs have been implicated in most cases.
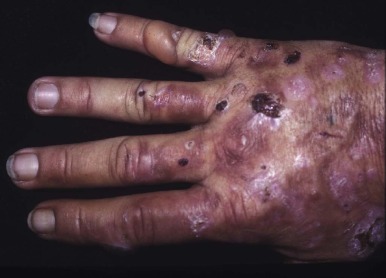
The cutaneous manifestations of HEP can be very dramatic and may resemble those seen in CEP. Recurrent blisters and erosions complicated by infection can lead to severe scarring, scleroderma-like changes, and photomutilation. In other cases the skin is less severely damaged and appears more like mild PCT. Hemolytic anemia and splenomegaly may be present, findings not associated with PCT.
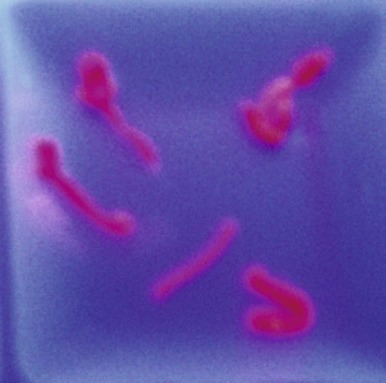
Evidence of abnormal liver function is nearly always present in patients with PCT, even in those without hepatitis C. In some reports, approximately 80% of males with PCT have been infected with hepatitis C but the association of hepatitis C with PCT is lower in northern Europe. The high concentration of porphyrins in the liver results in brilliant fluorescence of liver biopsy samples illuminated with a Wood’s light ( Fig. 12-15 ). Microscopic examination reveals needle-shaped, fluorescent porphyrin crystals in hepatocytes. Other histologic findings are not specific, but increased hepatocellular iron stores are a nearly constant finding.
Pathogenesis.
Over 100 URO-D mutations have been identified in patients with PCT. The functional consequences of many of these mutations have been studied, but heterozygosity for mutant URO-D alleles alone cannot explain the phenotype, as half-normal URO-D activity is not rate-limiting. Immunoreactive URO-D in the liver of patients with clinically evident F-PCT is half-normal, yet enzyme activity is 25% of normal or less. No URO-D mutations are found in patients with S-PCT and immunoreactive URO-D in all tissues is normal. When S-PCT becomes clinically evident, immunoreactive URO-D in the liver remains normal, yet enzyme activity is reduced to approximately 25% of normal, the same level of activity found in F-PCT. These findings indicate that the specific activity of hepatic URO-D is affected. Furthermore, catalytic activity of URO-D returns to normal after phlebotomy-induced iron depletion, suggesting that an iron-dependent process is responsible for the reduction in the specific activity of hepatic URO-D. Similar findings, a reduction in URO-D activity without a change in URO-D protein, have been noted in several animal models of PCT. Collectively, these data suggest a common mechanism in the pathogenesis of PCT, namely generation of an inhibitor of URO-D by an iron-dependent mechanism.
The inhibitor of URO-D in humans with PCT and in animal models of PCT has been identified as uroporphomethene. Uroporphomethene differs from uroporphyrinogen due to oxidation of a single bridge carbon between adjacent pyrrole rings. Uroporphomethene is sufficiently flexible to adapt to the spatial requirements of the active site of URO-D, but the variance from the structure of uroporphyrinogen prevents the inhibitor from serving as a substrate.
Generation of the uroporphomethene inhibitor likely requires liver-specific isozymes of cytochrome P450, but the mechanism of the iron effect remains unidentified. Iron clearly plays a central role because iron depletion through phlebotomy therapy reverses the clinical and biochemical phenotype, even when liver iron content is not clearly elevated. Replacement of iron removed through phlebotomy therapy produces a prompt relapse. Iron depletion prevents or dramatically attenuates the porphyric phenotype in animal models of PCT, and iron supplementation accelerates development of the phenotype. Hepcidin, a peptide that regulates systemic iron homeostasis, has been studied in patients with conflicting results. In one study, hepatic expression of hepcidin mRNA was significantly reduced, but in another study, serum hepcidin levels were increased. In spite of these data, very few patients with hemochromatosis or other disorders associated with iron overload develop PCT. Hepatic iron loading, therefore, is not sufficient to produce the PCT phenotype. Other factors, both genetic and environmental, appear to be required to create susceptibility to PCT.
The cutaneous findings in PCT, as in other porphyrias associated with photosensitivity, result from interaction of light with a wavelength of approximately 400 nm (Soret band) with porphyrins that accumulate in the skin. The photoexcited porphyrin returns to a stable state by emitting energy as a photon that disrupts membranes and intracellular organelles. The photodynamic reaction activates inflammatory mediators, which damage the epidermal basement membrane, causing fragility of the skin and blistering. Another prominent site of photodamage is at the vascular level of the skin, where thickening of superficial dermal vessel walls is a constant finding.
Laboratory Diagnosis.
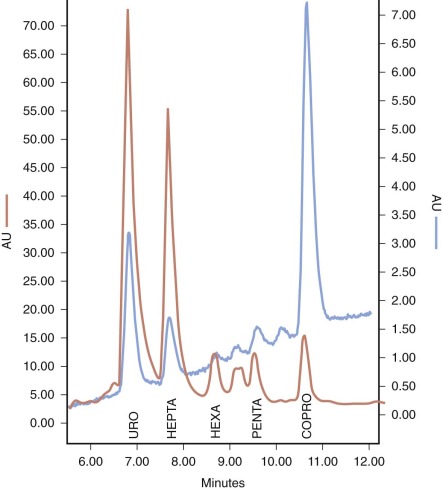
Laboratory testing is essential for confirmation of the clinical diagnosis of PCT and HEP. The appearance of the skin and skin biopsy findings resemble those of other photosensitizing porphyrias and pseudoporphyria. A useful initial approach to establishing that a porphyric disorder is present is to measure the total porphyrin content of plasma. A normal value excludes the diagnosis of PCT and HEP. If the plasma porphyrin level is high, additional testing is required to distinguish PCT and HEP from HCP and VP. Chromatographic separation and quantification of urinary and stool porphyrins serves this purpose. In PCT and HEP, greatly increased amounts of uroporphyrin and heptacarboxyl porphyrin are excreted in the urine ( Fig. 12-16 ). Urinary excretion of coproporphyrin may be slightly increased. Values for daily urine porphyrin excretion average 3000 µg (normal is less than 50 µg) and may be considerably higher. Cutaneous symptoms are usually mild or absent when uroporphyrin excretion is less than 1000 µg/day. A distinctive tetracarboxylic porphyrin, isocoproporphyrin, is excreted in feces, but total fecal porphyrin excretion is normal or minimally increased. HEP can be distinguished from PCT by the presence of anemia and an increase in erythrocyte zinc protoporphyrin.
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


