Recently, the development of poly(adenosine diphosphate–ribose) polymerase (PARP) inhibitors as a synthetic lethality approach has brought a major breakthrough in the treatment of breast cancer susceptibility gene (BRCA)-mutant cancers. Because sporadic cancers have also been found to commonly have other defects in DNA repair, PARP inhibitors are under active clinical investigation in combination with DNA-damaging therapeutics in a wide range of sporadic cancers. In this review, the authors discuss DNA repair mechanisms and PARP as a therapeutic target and summarize an update on clinical trials of available PARP inhibitors and predictive biomarkers for their efficacy.
- •
Many mechanisms are present in a cell to repair DNA damage, which when effective allow for cell survival. Understanding these mechanisms are important for both therapeutic treatment of cancer and overcoming resistance.
- •
The concept of synthetic lethality has be tested in treating patients with BRCA mutation with a PARP inhibitor. Initial results with olaparib was encouraging and supportive of synthetic lethality principle.
- •
The role of PARP inhibitor outside of BRCA mutation tumors await confirmation. Multiple studies are ongoing investigating PARP inhibitor with chemotherapy to see if inhibiting DNA repair will lead to tumor reduction and ultimately benefit to the patients.
DNA damage and repair mechanisms
DNA damage is generated by a variety of factors, not only from external factors such as chemical agents, UV light, or ionizing radiation but also from internal factors such as reactive oxygen species or intrinsic DNA replication errors during cell division. DNA damages, if let unrepaired, can cause errors of DNA synthesis during replication, leading to cell death or irreversible mutations resulting in long-term oncogenesis. Thus, individuals with an inherited defect in the DNA repair system are often at an increased risk of cancer. Common errors include (1) base modifications by frequently reactive oxygen species or chemical agents, such as loss of an amino group (deamination) or alkylation; (2) mismatch of nucleotide pairs by replication errors; (3) single-strand break (SSB) or double-strand break (DSB) by ionizing radiation; (4) failures in normal DNA metabolism by topoisomerases and nuclease; or (5) cross-links, covalent linkages between bases on intrastrand or interstrand. DSBs are the most critical form of DNA damage and can result in problems for transcription, replication, and chromosome segregation, eventually leading to apoptosis or carcinogenesis.
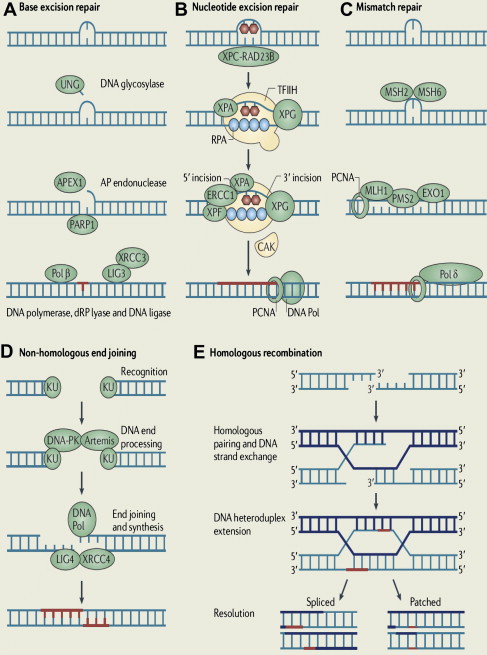
To maintain genetic stability against constantly occurring DNA lesions, several strategies of DNA damage detection and repair have evolved. The principal and partly overlapping DNA repair pathways in humans can be largely divided into 2 groups: one is the repair pathway for DNA SSB and the other is for DNA DSB. The former can be subdivided into 3 different repair processes: (1) repair of base damage and SSBs by base excision repair (BER), (2) repair of bulky DNA adducts by nucleotide excision repair (NER), and (3) repair of mismatches and insertion/deletion loops by DNA mismatch repair (MMR). The DSB is repaired through (1) homologous recombination (HR) and (2) nonhomologous end joining (NHEJ).
Base Excision Repair
The BER pathway repairs damage to a single or a few bases caused by reactive oxygen species, alkylating agents, or ionizing radiation, such as oxidation, alkylation, hydrolysis, or deamination, that could cause mutations by incorrect base pairing or lead to breaks in DNA during replication if uncorrected ( Fig. 1 A). PARP1 and PARP2 are recruited to the site of the SSB in this pathway.
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


