Recently, the development of poly(adenosine diphosphate–ribose) polymerase (PARP) inhibitors as a synthetic lethality approach has brought a major breakthrough in the treatment of breast cancer susceptibility gene (BRCA)-mutant cancers. Because sporadic cancers have also been found to commonly have other defects in DNA repair, PARP inhibitors are under active clinical investigation in combination with DNA-damaging therapeutics in a wide range of sporadic cancers. In this review, the authors discuss DNA repair mechanisms and PARP as a therapeutic target and summarize an update on clinical trials of available PARP inhibitors and predictive biomarkers for their efficacy.
- •
Many mechanisms are present in a cell to repair DNA damage, which when effective allow for cell survival. Understanding these mechanisms are important for both therapeutic treatment of cancer and overcoming resistance.
- •
The concept of synthetic lethality has be tested in treating patients with BRCA mutation with a PARP inhibitor. Initial results with olaparib was encouraging and supportive of synthetic lethality principle.
- •
The role of PARP inhibitor outside of BRCA mutation tumors await confirmation. Multiple studies are ongoing investigating PARP inhibitor with chemotherapy to see if inhibiting DNA repair will lead to tumor reduction and ultimately benefit to the patients.
DNA damage and repair mechanisms
DNA damage is generated by a variety of factors, not only from external factors such as chemical agents, UV light, or ionizing radiation but also from internal factors such as reactive oxygen species or intrinsic DNA replication errors during cell division. DNA damages, if let unrepaired, can cause errors of DNA synthesis during replication, leading to cell death or irreversible mutations resulting in long-term oncogenesis. Thus, individuals with an inherited defect in the DNA repair system are often at an increased risk of cancer. Common errors include (1) base modifications by frequently reactive oxygen species or chemical agents, such as loss of an amino group (deamination) or alkylation; (2) mismatch of nucleotide pairs by replication errors; (3) single-strand break (SSB) or double-strand break (DSB) by ionizing radiation; (4) failures in normal DNA metabolism by topoisomerases and nuclease; or (5) cross-links, covalent linkages between bases on intrastrand or interstrand. DSBs are the most critical form of DNA damage and can result in problems for transcription, replication, and chromosome segregation, eventually leading to apoptosis or carcinogenesis.
To maintain genetic stability against constantly occurring DNA lesions, several strategies of DNA damage detection and repair have evolved. The principal and partly overlapping DNA repair pathways in humans can be largely divided into 2 groups: one is the repair pathway for DNA SSB and the other is for DNA DSB. The former can be subdivided into 3 different repair processes: (1) repair of base damage and SSBs by base excision repair (BER), (2) repair of bulky DNA adducts by nucleotide excision repair (NER), and (3) repair of mismatches and insertion/deletion loops by DNA mismatch repair (MMR). The DSB is repaired through (1) homologous recombination (HR) and (2) nonhomologous end joining (NHEJ).
Base Excision Repair
The BER pathway repairs damage to a single or a few bases caused by reactive oxygen species, alkylating agents, or ionizing radiation, such as oxidation, alkylation, hydrolysis, or deamination, that could cause mutations by incorrect base pairing or lead to breaks in DNA during replication if uncorrected ( Fig. 1 A). PARP1 and PARP2 are recruited to the site of the SSB in this pathway.
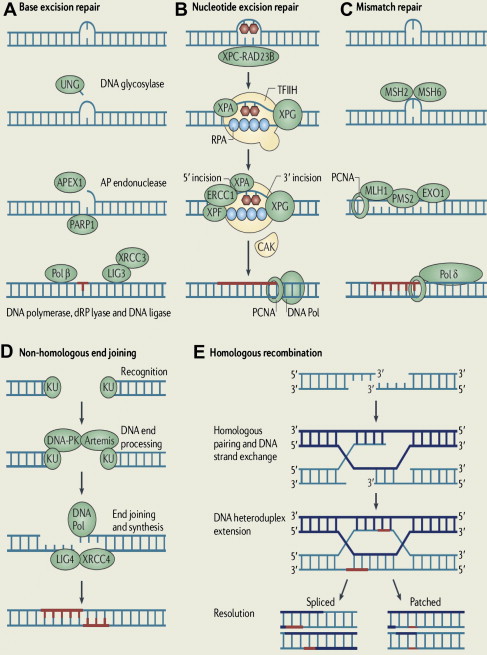
Nucleotide Excision Repair
NER is used for the removal of large patches around DNA lesions that are bulky and/or alter the helical structure of the DNA molecule, including those caused by UV radiation, carcinogenic compounds, and cross-linking chemotherapeutic agents (see Fig. 1 B).
Mismatch Repair
MMR removes base mismatches by DNA polymerases and small insertion-deletion loops introduced during DNA replication and recombination (see Fig. 1 C). A variety of base pair abnormalities resulting from DNA damage are also subject to processing by MMR, which include base pairs containing O 6-methylguanine, carcinogen adducts, UV photo products, and cisplatin adducts. Defects in the MMR system dramatically increase mutation rates resulting in hereditary nonpolyposis colorectal cancer and some types of sporadic tumor.
Nonhomologous End Joining
NHEJ can occur throughout the cell cycle, predominantly in the G1 phases. DNA break ends are directly ligated without the use of a homologous template, so NHEJ is prone to introducing errors ranging from small insertions and deletions at the break site to the joining of previously unlinked DNA ends (see Fig. 1 D).
Homologous Recombination
HR repairs DSB by using the undamaged sister chromatid or homologous chromosome, resulting in error-free repair, and occurs primarily during the S and G2 phases of the cell cycle. BRCA proteins are involved in this pathway (see Fig. 1 E).
DNA repair synthetic lethality as a therapeutic target
PARP in DNA Repair
PARP is an abundant nuclear enzyme involved in several cellular processes involving mainly DNA repair and programmed cell death. PARP1 and PARP2 are the only members of the PARP family that are known to be involved in the DNA SSB repair via the BER pathway. Once PARP detects an SSB, it binds to the sites of DNA damage through its DNA-binding domain and begins the synthesis of a poly(ADP-ribose) (PAR) chain from the substrate nicotinamide adenine dinucleotide (NAD + ) at sites of breakage. PARP induces poly(ADP-ribosyl)ation in PARP itself at the automodification domain and other proteins such as histone (H1 and H2B), which leads to chromatin decondensation to accommodate various DNA repair enzymes. The auto-poly(ADP-ribosyl)ation of PARP1 mediates the recruitment of BER proteins such as DNA polymerase β, DNA ligase III, and scaffolding proteins such as x-ray repair cross-complementing 1 (XRCC1) heterodimer. PARP1 then dissociates from the DNA because of its negative charge resulting from poly(ADP-ribosyl)ation, and the PAR chains are degraded by PAR glycohydrolase and possibly the ADP-ribose hydrolase ARH3 after repair of the DNA break. Besides its firmly established role in BER and DNA SSB repair, PARP1 is involved in multiple types of other DNA damage repair processes, including the repair of DNA DSBs, DNA cross-links, and stalled replication forks. PARP1 promotes restart of stalled replication forks and HR by recruiting HR factors including ATM, MRE11, and NBS1. Furthermore, PARP1 is recruited for DSB repair in the absence of essential components of the classical pathway of NHEJ such as Ku70. Although PARP1 is the predominant enzyme that synthesizes PAR in response to DNA damage, PARP2 has been shown to interact with PARP1 and is also implicated in BER. PARP2 is, however, unable to fully compensate for the loss of PARP1, accounting for approximately 10% of the total PARP activity of human cells.
BRCA1 and BRCA2 in DNA Repair
BRCA1 plays a key role in the regulation of the cell cycle checkpoints and the HR-mediated DNA repair pathway. On DNA damage, BRCA1 is rapidly phosphorylated and redistributes to sites of DNA breaks, where it interacts with RAD51 and other proteins involved in DNA DSB repair, such as MRN and CtIP. Although BRCA1 appears to function upstream of RAD51 filament polymerization, BRCA2 directly binds to and translocates RAD51 to areas of DNA damage and stabilizes RAD51 filaments on DSB. Absence of either factor causes DNA repair defect, primarily in HR, which contributes to tumorigenesis. Heterozygous germline mutations in BRCA1 or BRCA2 predispose to various type of cancers, especially breast and ovarian cancers. This HR deficiency also has a critical impact on chemosensitivity. BRCA1 -mutated breast cancer cells are highly sensitive to DNA interstrand cross-linking–inducing agents such as cisplatin and mitomycin C because they disrupt replication forks during S phase, which requires HR-mediated DSB repair for S phase progression and cell survival. Because of their heightened cytotoxicity in HR-defective cells, platinum agents have been extensively applied as chemotherapeutic drugs in BRCA1 – or BRCA2 -associated cancers with outcomes better than those of non- BRCA -associated cancers.
Synthetic Lethality
The term synthetic lethality was coined in 1946. It describes a phenomenon in which 2 nonlethal mutations have no effect on cell viability in the presence of either mutation alone but lead to cell death in combination. This concept is now being exploited in cancer treatment. Because many cancer cells have cancer-relevant genetic lesions that are not present in normal cells, mimicking the effect of a second genetic mutation with a targeted agent should selectively kill only cancer cells with a large therapeutic window. To date, the most successful synthetic lethality relationship in DNA repair pathways for cancer treatment comes from PARP inhibition in BRCA -null tumors. Although PARP1 plays a critical role in DNA SSB repair, loss of PARP1 activity is not lethal in normal cells. When unrepaired DNA SSBs caused by the absence of PARP1 activity encounter DNA replication forks, they result in stalled replication forks that are subsequently converted to DNA DSBs. Although these DNA DSBs are effectively repaired by the HR pathway in normal cells, cells that are defective in HR, such as BRCA1 – or BRCA2 -deficient cells, cannot repair them, resulting in cell death. Therefore, PARP inhibitors could be selectively lethal to cells lacking functional BRCA1 or BRCA2 with minimal toxicity to normal cells.
Synthetic lethality in BRCA1 – and BRCA2 -deficient cells when exposed to PARP inhibitors was confirmed in in vitro and in vivo mouse models. Farmer and colleagues showed that PARP1 depletion with RNA interference decreased clonogenic survival of BRCA1 – and BRCA2 -deficient embryonic stem cells compared with wild-type cells. Furthermore, clonogenic cell survival assays showed that BRCA1 – or BRCA2 -deficient cells were much more sensitive to the potent PARP inhibitors, KU0058684 and KU0058948, than heterozygous mutant or wild-type cells. KU0058684 also blocked tumor growth in vivo in BRCA2 -deficient cells. Similarly, Bryant and colleagues demonstrated that BRCA2 -deficient V-C8 cells were profoundly sensitive to the PARP inhibitors NU1025 and AG014361, as compared with the wild-type V79 cells. Human breast cancer cell lines MCF-7 and MDA-MB-231 also displayed a similar sensitivity to NU1025 on depletion of BRCA2 with RNA interference.
BRCAness
The primary challenge to the promise of PARP inhibitors in the treatment of BRCA -mutant cancers is the low frequency of germline mutations in BRCA1 or BRCA2 , only 10% to 15% of unselected ovarian cancers and 5% to 10% of breast cancers. In addition, BRCA1 and BRCA2 genes are infrequently mutated somatically in sporadic cancers, with 4% to 9% of unselected or sporadic ovarian cancers. However, some sporadic cancers phenotypically behave like BRCA1/2 -mutant cancers even though they do not have known BRCA mutations. These tumors may carry abnormalities in the expression or function of BRCA1 or BRCA2 or in other critical components of the HR DNA repair pathway. This phenomenon is called BRCAness and is characterized by defective HR. One of the mechanisms of BRCAness is the silencing of BRCA1 or BRCA2 through the aberrant methylation of the promoter and has been reported in 11% to 14% of sporadic breast cancers and 5% to 18% of ovarian cancers. Preclinical studies have shown that tumor cell lines with decreased BRCA expression by mutation or epigenetic silencing responded equally well to the PARP inhibitor AG014699 that additionally inhibited the growth of epigenetically silenced BRCA1 xenograft tumors. These suggest that PARP inhibitors may have a potential role in sporadic cancers as well as hereditary cancers. The amplification of the EMSY gene that encodes for EMSY protein that can interact with and silence the activation domain of BRCA2 has also been described as a potential mechanism of BRCA inactivation. The amplification of the EMSY gene was reported in 13% of sporadic breast cancers and 17% of high-grade serous ovarian cancers (HGSOCs). Because Fanconi anemia proteins are involved in the HR pathway, the epigenetic silencing of Fanconi anemia complementation group F ( FANCF ) gene through promoter methylation is another potential mechanism of BRCAness. The inhibition of the FANCF gene in ovarian cancer cell lines through promoter methylation was associated with enhanced sensitivity to DNA-damaging agents such as cisplatin; in reverse, demethylation of the FANCF promoter resulted in cisplatin resistance. In addition to potential transcriptional and posttranslational abnormalities occurring in BRCA1 and BRCA2 pathways, deficiency of proteins involved in the HR pathway, such as RAD51, RAD54, DSS1, RPA1, NBS1, ATR, ATM, CHK1, or CHK2, can also induce sensitivity to PARP inhibitors.
DNA repair synthetic lethality as a therapeutic target
PARP in DNA Repair
PARP is an abundant nuclear enzyme involved in several cellular processes involving mainly DNA repair and programmed cell death. PARP1 and PARP2 are the only members of the PARP family that are known to be involved in the DNA SSB repair via the BER pathway. Once PARP detects an SSB, it binds to the sites of DNA damage through its DNA-binding domain and begins the synthesis of a poly(ADP-ribose) (PAR) chain from the substrate nicotinamide adenine dinucleotide (NAD + ) at sites of breakage. PARP induces poly(ADP-ribosyl)ation in PARP itself at the automodification domain and other proteins such as histone (H1 and H2B), which leads to chromatin decondensation to accommodate various DNA repair enzymes. The auto-poly(ADP-ribosyl)ation of PARP1 mediates the recruitment of BER proteins such as DNA polymerase β, DNA ligase III, and scaffolding proteins such as x-ray repair cross-complementing 1 (XRCC1) heterodimer. PARP1 then dissociates from the DNA because of its negative charge resulting from poly(ADP-ribosyl)ation, and the PAR chains are degraded by PAR glycohydrolase and possibly the ADP-ribose hydrolase ARH3 after repair of the DNA break. Besides its firmly established role in BER and DNA SSB repair, PARP1 is involved in multiple types of other DNA damage repair processes, including the repair of DNA DSBs, DNA cross-links, and stalled replication forks. PARP1 promotes restart of stalled replication forks and HR by recruiting HR factors including ATM, MRE11, and NBS1. Furthermore, PARP1 is recruited for DSB repair in the absence of essential components of the classical pathway of NHEJ such as Ku70. Although PARP1 is the predominant enzyme that synthesizes PAR in response to DNA damage, PARP2 has been shown to interact with PARP1 and is also implicated in BER. PARP2 is, however, unable to fully compensate for the loss of PARP1, accounting for approximately 10% of the total PARP activity of human cells.
BRCA1 and BRCA2 in DNA Repair
BRCA1 plays a key role in the regulation of the cell cycle checkpoints and the HR-mediated DNA repair pathway. On DNA damage, BRCA1 is rapidly phosphorylated and redistributes to sites of DNA breaks, where it interacts with RAD51 and other proteins involved in DNA DSB repair, such as MRN and CtIP. Although BRCA1 appears to function upstream of RAD51 filament polymerization, BRCA2 directly binds to and translocates RAD51 to areas of DNA damage and stabilizes RAD51 filaments on DSB. Absence of either factor causes DNA repair defect, primarily in HR, which contributes to tumorigenesis. Heterozygous germline mutations in BRCA1 or BRCA2 predispose to various type of cancers, especially breast and ovarian cancers. This HR deficiency also has a critical impact on chemosensitivity. BRCA1 -mutated breast cancer cells are highly sensitive to DNA interstrand cross-linking–inducing agents such as cisplatin and mitomycin C because they disrupt replication forks during S phase, which requires HR-mediated DSB repair for S phase progression and cell survival. Because of their heightened cytotoxicity in HR-defective cells, platinum agents have been extensively applied as chemotherapeutic drugs in BRCA1 – or BRCA2 -associated cancers with outcomes better than those of non- BRCA -associated cancers.
Synthetic Lethality
The term synthetic lethality was coined in 1946. It describes a phenomenon in which 2 nonlethal mutations have no effect on cell viability in the presence of either mutation alone but lead to cell death in combination. This concept is now being exploited in cancer treatment. Because many cancer cells have cancer-relevant genetic lesions that are not present in normal cells, mimicking the effect of a second genetic mutation with a targeted agent should selectively kill only cancer cells with a large therapeutic window. To date, the most successful synthetic lethality relationship in DNA repair pathways for cancer treatment comes from PARP inhibition in BRCA -null tumors. Although PARP1 plays a critical role in DNA SSB repair, loss of PARP1 activity is not lethal in normal cells. When unrepaired DNA SSBs caused by the absence of PARP1 activity encounter DNA replication forks, they result in stalled replication forks that are subsequently converted to DNA DSBs. Although these DNA DSBs are effectively repaired by the HR pathway in normal cells, cells that are defective in HR, such as BRCA1 – or BRCA2 -deficient cells, cannot repair them, resulting in cell death. Therefore, PARP inhibitors could be selectively lethal to cells lacking functional BRCA1 or BRCA2 with minimal toxicity to normal cells.
Synthetic lethality in BRCA1 – and BRCA2 -deficient cells when exposed to PARP inhibitors was confirmed in in vitro and in vivo mouse models. Farmer and colleagues showed that PARP1 depletion with RNA interference decreased clonogenic survival of BRCA1 – and BRCA2 -deficient embryonic stem cells compared with wild-type cells. Furthermore, clonogenic cell survival assays showed that BRCA1 – or BRCA2 -deficient cells were much more sensitive to the potent PARP inhibitors, KU0058684 and KU0058948, than heterozygous mutant or wild-type cells. KU0058684 also blocked tumor growth in vivo in BRCA2 -deficient cells. Similarly, Bryant and colleagues demonstrated that BRCA2 -deficient V-C8 cells were profoundly sensitive to the PARP inhibitors NU1025 and AG014361, as compared with the wild-type V79 cells. Human breast cancer cell lines MCF-7 and MDA-MB-231 also displayed a similar sensitivity to NU1025 on depletion of BRCA2 with RNA interference.
BRCAness
The primary challenge to the promise of PARP inhibitors in the treatment of BRCA -mutant cancers is the low frequency of germline mutations in BRCA1 or BRCA2 , only 10% to 15% of unselected ovarian cancers and 5% to 10% of breast cancers. In addition, BRCA1 and BRCA2 genes are infrequently mutated somatically in sporadic cancers, with 4% to 9% of unselected or sporadic ovarian cancers. However, some sporadic cancers phenotypically behave like BRCA1/2 -mutant cancers even though they do not have known BRCA mutations. These tumors may carry abnormalities in the expression or function of BRCA1 or BRCA2 or in other critical components of the HR DNA repair pathway. This phenomenon is called BRCAness and is characterized by defective HR. One of the mechanisms of BRCAness is the silencing of BRCA1 or BRCA2 through the aberrant methylation of the promoter and has been reported in 11% to 14% of sporadic breast cancers and 5% to 18% of ovarian cancers. Preclinical studies have shown that tumor cell lines with decreased BRCA expression by mutation or epigenetic silencing responded equally well to the PARP inhibitor AG014699 that additionally inhibited the growth of epigenetically silenced BRCA1 xenograft tumors. These suggest that PARP inhibitors may have a potential role in sporadic cancers as well as hereditary cancers. The amplification of the EMSY gene that encodes for EMSY protein that can interact with and silence the activation domain of BRCA2 has also been described as a potential mechanism of BRCA inactivation. The amplification of the EMSY gene was reported in 13% of sporadic breast cancers and 17% of high-grade serous ovarian cancers (HGSOCs). Because Fanconi anemia proteins are involved in the HR pathway, the epigenetic silencing of Fanconi anemia complementation group F ( FANCF ) gene through promoter methylation is another potential mechanism of BRCAness. The inhibition of the FANCF gene in ovarian cancer cell lines through promoter methylation was associated with enhanced sensitivity to DNA-damaging agents such as cisplatin; in reverse, demethylation of the FANCF promoter resulted in cisplatin resistance. In addition to potential transcriptional and posttranslational abnormalities occurring in BRCA1 and BRCA2 pathways, deficiency of proteins involved in the HR pathway, such as RAD51, RAD54, DSS1, RPA1, NBS1, ATR, ATM, CHK1, or CHK2, can also induce sensitivity to PARP inhibitors.
PARP inhibitors
The profound sensitivity of BRCA -mutant cells to PARP inhibition prompted the development of PARP inhibitors for cancer therapy ( Table 1 ). At present, most PARP inhibitors in preclinical and clinical studies are third-generation PARP inhibitors and compete with the substrate NAD + for the catalytic domain of the PARP enzyme, leading to reversible inhibition of the enzyme. AZD2281/KU-0059436 (olaparib) and ABT-888 (veliparib) are the most common inhibitors assessed to date in clinical trials. Both drugs are administered orally and are potent inhibitors of PARP1 and PARP2, with a half maximal inhibitory concentration (IC 50 or K i ) in the nanomolar range for 2 enzymes. Preclinical activity of AG014361 has also been translated into the clinic as its clinical analogue CO338 (rucaparib, AG014699, PF-01367338) administered intravenously. It was initially tested in combination with temozolomide in melanoma. Other PARP inhibitors under active clinical investigation include MK4827, BMN 673, CEP-9722, and E7016/GPI 21016 (see Table 1 ). Of note, BSI-201 (iniparib) was initially described as having PARP inhibitory activity, which proceeded into a phase 3 trial in triple-negative breast cancer (TNBC) in combination with carboplatin and paclitaxel, but recent evidence suggests that iniparib does not seem to inhibit PARP1 and PARP2 at the clinical dose.
| Name | Route | Disease | Single/Combination Therapy | Phase of Clinical Development a | Company |
|---|---|---|---|---|---|
| Rucaparib (CO338, AGO 14699, PF-0367338) | IV | BRCA + ovarian/breast cancer | Single | 2 | Clovis Oncology (Boulder, CO, USA) |
| Melanoma | Temozolomide | 2 | |||
| Olaparib (AZD2281) | Oral | BRCA + ovarian cancer | Single agent | 2 | AstraZeneca (London, UK) |
| BRCA + breast cancer | |||||
| High-grade SOC/TNBC | |||||
| Various solid tumors | Carboplatin, topotecan, paclitaxel, dacarbazine, or gemcitabine/cisplatin | 1 | |||
| Veliparib (ABT-888) | Oral | Melanoma | Temozolomide | 2 | Abbott (Abbott Park, IL, USA) |
| Breast cancer | |||||
| Colorectal cancer | |||||
| Various solid tumors | Topotecan, irinotecan, cyclophosphamide, or doxorubicin/cyclophosphamide | 1 | |||
| MK 4827 | Oral | HGSOC enriched with BRCA + | Single | 1 | Merck (Whitehouse Station, NJ, USA) |
| BMN 673 | Oral | Various solid tumors Hematologic malignancies | Single agent | 1 | Biomarin (Novato, CA, USA) |
| CEP-9722 | Oral | Advanced solid tumor (phase 1)/cancer with defective DNA repair pathway (phase 2) | Single | 1/2 | Cephalon (Frazer, PA, USA) |
| Solid tumor | Temozolomide Gemcitabine/Cisplatin | 1 | |||
| E7016 (GPI 21016) | Oral | Solid tumor | Temozolomide | 1 | MGI Pharma/Eisai (Bloomington, MN, USA) |
a Indicates the most advanced phase in studies that have been published or presented. In CEP-9722 and E7016, ongoing trials are shown because there are no published studies.
Monotherapy with PARP inhibitors in BRCA -mutant cancers
On the basis of preclinical data showing hypersensitivity to PARP inhibitors in tumor cells lacking BRCA1 or BRCA2 , PARP inhibitor was used as a single agent in patients with BRCA mutation. The first landmark trial of PARP inhibitor as monotherapy was performed with olaparib enriched with BRCA1 / 2 mutation carriers and limited to patients with BRCA mutation ovarian cancer in the expansion phase. Olaparib was safe and well tolerated with no obvious differences in toxicities observed between BRCA1 or BRCA2 mutation carriers and noncarriers. The maximum tolerated dose (MTD) was 400 mg twice a day, and the dose-limiting toxicities (DLTs) included grade 3 mood alteration, fatigue, somnolence, and grade 4 thrombocytopenia. As expected, there was significant antitumor activity only in patients with BRCA1 – or BRCA2 -mutated cancers, including ovarian, breast, and prostate cancers, among whom 12 of 17 (63%) patients had clinical benefit and 9 of 19 (47%) patients achieved responses according to the Response Evaluation Criteria in Solid Tumors (RECIST). In the expansion cohort of 50 BRCA mutation carriers with ovarian, primary peritoneal, and fallopian tube cancers, 20 patients (40%) had a RECIST response, cancer antigen 125 responses, or both; 14 patients (28%) had RECIST responses. Pharmacodynamic assay demonstrated that a dose of 60 mg or more twice daily reduced PARP activity in peripheral blood mononuclear cells (PBMCs) by more than 90% as compared with the value at baseline. Plucked eyebrow hair follicles were collected before treatment and again 6 hours after treatment, showing induction of γ-H2AX, which did not seem to increase further at doses greater than 100 mg twice daily, the lowest dose of this analysis. Based on these results, a dose of 100 mg twice daily may be adequate for PARP inhibitory effect; this dose was further studied in phase 2 studies along with a twice-daily dose of 400 mg, the MTD.
Subsequently, 2 proof-of-concept phase 2 studies with olaparib in patients with advanced or recurrent breast (International Collaborative Expertise for BRCA Education and Research through Genetics [ICEBERG]1) or ovarian cancer (ICEBERG2) who had BRCA1 or BRCA2 mutations have provided further support for synthetic lethal approach to cancer therapy. Both studies recruited sequentially 2 dose cohorts treated with twice-daily doses of olaparib 400 mg (the established MTD) and then 100 mg (the lowest PARP inhibitory dose with clinical activity). In ICEBERG1 trial, the objective response rate in breast cancer seemed to be higher in the 400-mg cohort (41%, 11/27) than in the 100-mg cohort (22%, 6/27), and median progression-free survival (PFS) also seemed to be longer in the 400-mg cohort (5.7 vs 3.8 months). Similarly, ICEBERG2 trial in ovarian cancer showed that the high-dose cohort seemed to have better response rate (33% vs 13%) and median PFS (5.8 vs 1.9 months) than the low-dose cohort. Overall, both trials confirmed the tolerability of olaparib in BRCA 1 / 2 mutation carriers, with mainly grade 1 or 2 adverse events.
A subsequent randomized phase 2 study comparing the safety and efficacy of 2 doses of olaparib (200 or 400 mg twice daily continuously in 28-day cycles) with that of pegylated liposomal doxorubicin (PLD) (50 mg/m 2 every 4 weeks) was done in patients with BRCA mutation–associated ovarian cancer who had failed previous platinum-based chemotherapy. A total of 97 patients were randomized into 3 groups. Although the 400-mg group had a numerically longer median PFS (8.8 vs 6.5 vs 7.1 months, respectively) and a higher response rate (25% vs 18% vs 31%, respectively) than the 200-mg or PLD groups, neither outcome reached statistical significance. These negative outcomes might be partially attributed to better PFS in the PLD group compared with historical cohorts. Regarding safety, both doses of olaparib were well tolerated with less toxicity compared with PLD. The PLD group had more grade 3 or 4 toxicities. Although the primary end point was not fulfilled, this study demonstrated the consistent efficacy with favorable safety of olaparib as monotherapy in BRCA -mutant ovarian cancer.
Another promising PARP inhibitor that has been studied in BRCA1 / 2 mutation carriers is MK4827, a potent oral PARP1/2 inhibitor with an IC 50 of 3.8 and 2.1 nM for PARP1 and PARP2, respectively. A phase 1 study of MK4827 enriched with BRCA1 / 2 mutation carriers had established MTD at a dose of 300 mg once daily. DLTs included grade 3 fatigue and pneumonitis and grade 4 thrombocytopenia. Inhibition of PARP activity in PBMCs was demonstrated at doses of 80 mg or greater. The response rate was 37% (7/19) in patients with BRCA -mutated ovarian cancer and 20% (3/15) in patients with sporadic ovarian cancer. MK4827 was well tolerated with the most common toxicities of fatigue (52.5%), nausea (52.5%), vomiting (38.8%), diarrhea (21.3%), thrombocytopenia (33.8%), and neutropenia (21.3%), which were mostly graded 1 to 2 except for thrombocytopenia (grade 3/4, 15%).
Rucaparib was evaluated in a phase 2 study for BRCA1 / 2 -mutated advanced ovarian and/or breast cancer in which 18 mg/m 2 of rucaparib was given on days 1 to 5 of a 21-day cycle. PARP activity assessed in PBMC demonstrated a mean inhibition of 84% over baseline levels at 24 hours after a single dose of 18 mg/m 2 of AG014699. Although the overall response rate was 5% (2/38), 26% (10/38) achieved stable disease for 4 or more months. Rucaparib had an acceptable safety profile with drug-related toxicity, mainly grade 1 and 2; the most common toxicities were fatigue (39%), nausea (26.8%), diarrhea (19.5%), and dizziness (17.1%).
Overall, these data clearly provide clinical validation for single-agent PARP inhibitor activity in patients with BRCA1 / 2 mutations.
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


