Fig. 22.1
Simplified scheme of tumor progression. During progression, cancer cells recruit inflammatory cells and “educate” their phenotype (in the case of macrophages, from M1 to M2). The phenotype abbreviated as M2 contributes to angiogenic switch and formation of proangiogenic and immunosuppressive tumor microenvironment. The development of abnormal tumor vascular network and the resultant hypoxia lead to invasive tumor phenotype (invasive switch/epithelial-mesenchymal transition/EMT)
The aim of this chapter is to draw attention to the possibility of using such reprogrammed tumor microenvironment in anticancer therapeutic context.
22.2 Recruitment of Inflammatory Cells by Cancer Cells
Mutations of certain genes in cancer cells (e.g., RET, RAS, Myc, and p53) trigger transcription of genes encoding chemotactic factors, including CC and CXC subgroup chemokines, the main chemoattractants of inflammatory reaction [4].
Chemokines released by cancer cells lead to the recruitment of various inflammatory cells from the bloodstream. For example, monocytes and macrophages are recruited mainly by CCL2 chemokine, whereas dendritc cells (DCs) are recruited by CCL20. The recruitment of regulatory lymphocytes (Tregs) is accomplished by CCL22, while CXCL1, CXCL5, CXCL6, and CXCL8 mobilize polymorphonuclear leukocytes (PMNs) [8].
Generally, recruitment of immune reaction cells is stimulated by cytokines and growth factors. For example, tumor-associated macrophages (TAMs) are mobilized (besides CCL2, CCL5, CCL7, CXCL8, and CXCL12 chemokines) by VEGF and PDGF cytokines as well as M-CSF [9].
Mobilization and recruitment processes also involve damage-associated molecular pattern (DAMP) molecules, especially high-mobility group box 1 (HMGB1) protein. HMGB1 is passively released from necrotized cancer cells, whereas actively, it is released from immune cells. HMGB1 stimulates neutrophils and monocytes to release proinflammatory cytokines [7]; it is also proangiogenic [10, 11].
Recruited inflammatory cells are specifically “educated” by cytokines, growth factors, and chemokines released by cancer cells [5]. In essence, such “education” results in the appearance of a specific type of inflammatory cells. Generally, such a reaction leads to the appearance of tumor growth-promoting phenotype. Among the best-studied types of cells affected by appearance of such phenotype are macrophages. Rise of tumor-promoting phenotype among macrophages is possible due to extraordinary plasticity of these cells [12].
22.3 Macrophage Plasticity: M1 and M2 Phenotypes
Depending on organ localization, macrophages differ in phenotype and transcriptional profiles. As mentioned, macrophages display great plasticity, i.e., phenotype-changing capability [13]. Under the influence of TLR ligands (including LPS) or IFN-γ, macrophages exhibit the so-called M1 phenotype (classical macrophage activation), whereas alternative activation (M2) takes place when macrophages are stimulated by IL-4/IL-13 [14]. Polarization of macrophages into M1 and M2 classes reflects classification of Th1/Th2 immune cells.
Macrophages with M1 phenotype release proinflammatory cytokines (e.g., TNF-α, IL-1, IL-6, IL-12, and IL-23) [14]. Owing to increased expression of class I and II MHC molecules, M1 macrophages are capable of antigen presentation. These cells stimulate arginine metabolism and production of nitric oxide (NO) and citrulline. Due to released NO and reactive oxygen species (ROS), M1 cells are cytotoxic. M1 macrophage phenotype is controlled by signal transducer and activator of transcription 1 (STAT1) and interferon-regulatory factor 5 (IRF5) [15]. Typical M1 phenotype is correlated with elevated levels of released IL-12 and IL-23 as well as low level of IL-10 (IL-12high, IL-23high, IL-10low) [13]. In the course of inflammation, polarization from M1 to M2 macrophages is observed. At present, several M2 cell subtypes have been identified: M2a, M2b, and M2c. M2a phenotype is stimulated by IL-4 or IL-13 cytokines, whereas M2b is stimulated by LPS, TLR, and IL-1ra receptor antagonist, respectively. Finally, M2c phenotype is induced by IL-10, TGF-β, and glucocorticoids [14]. If M1 cells are capable of removing pathogen factors, then cells of M2 phenotype participate in reconstruction of damaged tissues and de novo formation of blood microvessels [13]. M2 macrophages do not release nitric oxide. Instead, they highly express arginase I which metabolizes arginine to ornithine and polyamines, compounds necessary to synthesize collagen and to subsequent fibrosis and damaged tissue remodeling. M2 cells feature high levels of mannose and galactose receptors, as well as scavenger receptors. M2 inhibits the release of CXCL5, CXCL9, and CXCL10 chemokines while stimulating that of CCL24, CCL17, and CCL22, which facilitate the recruitment of eosinophils, basophils, and Th2 cells. Due to released IL-10 cytokine, M2 macrophages become immunosuppressive. M2 phenotype is controlled by STAT6, IRF4, and peroxisome proliferator-activated receptor-γ (PPARγ) [15]. Typical phenotype of M2 cells is linked with low levels of IL-12 and IL-23 and high level of IL-10 (IL-12low, IL-23 low, IL-10 high) [13].
22.4 TAM: Cells with M2 Phenotype
In tumors, a specific population of macrophages (tumor-associated macrophages (TAMs)) has been observed. TAM cells make up for over 50 % of the tumor mass [9]. The phenotype of TAMs is similar to that of M2 macrophages [16]. Formation of this phenotype from progenitors requires Th2 lymphocytes (which are the source of IL-4 and IL-13), Treg lymphocytes (which synthesize TGF-β and IL-10), as well as cancer cells and tumor-specific fibroblasts (CAFs).
TAMs play different roles in tumor environment [17]. They strongly affect tumor progression. For example, TAMs synthesize EGF, which stimulates the growth of cancer cells. They release proangiogenic factors (VEGF, PDGF, and TGF-β) and several FGF family factors. TAMs stimulate immunosuppression (IL-10) [6]. Through the release of CCL17 and CCL22 chemokines, TAMs are capable of recruiting T lymphocytes (Treg and Th2). TAM cells also release CCL8, which recruits “naïve” T lymphocytes. These lymphocytes become anergic in the tumor microenvironment.
TAM macrophages have the tendency to accumulate in underoxygenated (hypoxic) tumor regions [17]. Under such conditions, TAMs induce transcription factor HIF-1α, VEGF, and CXCL12 (and its receptor CXCR4), which modulate TAM migration into avascular regions. HIF-1α controls expression of inducible nitric oxide synthase (iNOS) and arginase 1 (Arg1). At low concentrations of IFN-γ, transcription factor HIF-2α induces expression of Arg1, inhibits NO synthesis, and favors formation of Th2 phenotype. Under high IFN-γ concentration, HIF-1α dominates. The latter stimulates induction of iNOS, which metabolizes arginine to NO and leads to the appearance of Th1 phenotype [5].
TAM cells release immunosuppressive cytokines (TGF-β and IL-10) and synthesize the immunosuppressive arginase 1 enzyme [14]. These cytokines and arginase exert considerable effects on the growth of cancer cells. TGF-β cytokine stimulates M1 to M2 polarization of macrophages and inhibits cytolytic activity of NK cells, as well as migration and activity of DCs. TGF-β stimulates differentiation of CD4+T cells to Th2 and blocks activity of CD8+ T cells by inhibiting the activity of granzyme A and B as well as IFN-γ. TGF-β also promotes the activity of Treg lymphocytes.
Immunosuppressive interleukin-10 is released by both TAM and CD8+ cells, as well as by cancer cells. IL-10 inhibits the activity of IL-12, maturation of DCs, and release of cytotoxic cytokine IFN-γ, the main cytokine stimulating differentiation of “naïve” T lymphocytes [6, 14].
Arginase 1, a Th2 cell molecular marker, is also active in cancer cells. This enzyme exerts a negative effect on functioning of T cell receptors (TCRs) and inhibits CD8+ T cell response [14].
TAM cells are programmed to release proangiogenic factors and enzymes involved in the formation of blood vasculature [6, 14]. Proangiogenic agents include, among others, VEGF, PDGF, TGF-β, and FGF, whereas enzymes modifying extracellular matrix (ECM) are MMP-2, MMP-7, MMP-9, MMP-12, and “plasmin system”. MMP-9 metalloproteinase releases proangiogenic factors sequestered by extracellular matrix proteins [6]. TAM macrophages also participate in the formation of vascular junctions [18] and play a major role in the creation of the so-called angiogenic switch [19]. As a result of this switch, tumors shift from avascular type of growth to vascular one (and become dependent on the formation of own blood vascular supply). TAM cells which synthesize VEGF-C and VEGF-D also participate in the formation of lymphatic vessels [6].
TAM cells, as well as other immune cells (e.g., Treg lymphocytes), link immunosuppression and angiogenesis [20–23]. This link between angiogenesis and immunosuppression may be due to pluripotent properties of some proangiogenic agents. VEGF is not only proangiogenic, it also acts as an inhibitor of dendritic cells’ maturation [24]. VEGF also stimulates proliferation of Treg lymphocytes [23]. PlGF, VEGFR1 receptor ligand also inhibits differentiation of DCs [25]. HGF factor inhibits antigen presentation by DCs and stimulates the appearance of Th2 lymphocytes [26]. TGF-β is not only proangiogenic but also a cytokine which stimulates polarization of M1 macrophages into M2 [27].
Macrophages possess a dual nature (thus they have been dubbed “a double-edged sword”): under certain conditions, they are cytotoxic and eliminate cancer cells (e.g., M1 macrophages), while under others, they stimulate tumor growth being proangiogenic and immunosuppressive (e.g., TAM (M2) macrophages) [28].
Polarization of macrophages depends on the environmental context of various signals secreted by both cancer and other tumor milieu cells [13]. Depending on certain signals’ domination, macrophage cells present either M1 or M2 phenotype. Domination of IFN-γ results in the appearance of M1 phenotype. On the other hand, IL-4/IL-13 and TGF-β in tumor microenvironment induce M2 phenotype (TAM) in macrophage cells. Under hypoxic conditions, macrophages display their M2 phenotype.
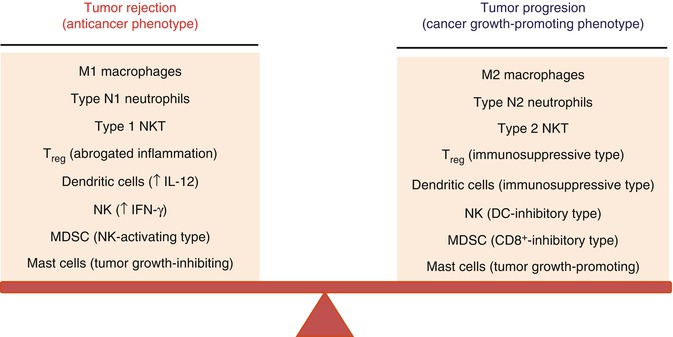
Dual (bipolar) phenotypes are exhibited also by other cells of the immune system. Depending on circumstances, such cells display a phenotype that either inhibits tumor growth or stimulates it [7]. For example, the presence of TGF-β, a strong immunosuppressant and proangiogenic factor, in tumor milieu results in tumor-associated neutrophils (TANs) becoming cells that stimulate tumor growth (type II) [29]. Milieu lacking TGF-β causes neutrophils to participate in the elimination of cancer cells (type I). Dual nature is also shown by NKT cells [30], DCs [31], mast cells [32], Treg cells [33], and NK cells [34, 35]. Figure 22.2 shows examples of immune system cells possessing such dual nature.
22.5 M1 → M2 Tumor Microenvironment Reversal: Therapeutic Approach
Is it possible to revert macrophage phenotype from M2 to M1? In other words, is it possible to revert tumor milieu from proangiogenic and immunosuppressive to anti-angiogenic and immunostimulatory one?
If M1 or M2 phenotype is a result of destabilized equilibrium between proangiogenic/immunosuppressive agents and anti-angiogenic/immunostimulatory ones, then shifting this equilibrium in favor of anti-angiogenic and immunostimulatory one (M1 phenotype), we can perhaps revert tumor growth dynamics and create conditions leading to elimination of cancer cells (Fig. 22.2).
One of the first therapeutic attempts of this kind was proposed by Guiducci et al. [39]. In order to revert M2 to M1 phenotype, these authors used an antibody-inhibiting IL-10R receptor and CpG oligonucleotide-activating TLR9 receptor. Combination of these drugs permitted a specific “reeducation” of M2 macrophages and led to a considerable therapeutic benefit. Specific “reeducation” of macrophages can also be accomplished by using other therapeutic agents. For instance, a CD40-activating agent (CD40 is a protein belonging to the TNF-like receptor superfamily) was shown to promote M2 → M1 conversion [40]. Therapeutic effects were also observed in animals burdened with tumors in which genes encoding NF-κB transcription factors were active [41] Also, certain proinflammatory agents (e.g., imiquimod), which stimulate TLR7/8 receptors, induce antitumor reaction. M2 → M1 conversion could also be observed in the case of polyI:C therapeutic agent (dsRNA analog) [42]. Its receptor turned out to be the Toll-like receptor 3 activating TICAM-1 signaling pathway. This pathway is not only important from the standpoint of dendritic cells’ maturation; it is also necessary for eliciting an antitumor response. A substantial role in M2 → M1 conversion is played by CpG oligonucleotides activating TLR9 receptors [43]. These factors inhibit immunosuppressive activity of monocytes and stimulate release of Th1 cytokine as well as the appearance of macrophages having cytotoxic properties (M1). Rolny et al. [44] demonstrated the effect of histidine-rich glycoprotein on normalization of blood vasculature and polarization of TAMs.
Different combinations of anti-angiogenics and immunostimulants also possess therapeutic properties [45, 46]. Sunitinib, a specific inhibitor of tyrosine kinase activity (found in many receptors of proangiogenic factors), when combined with IL-12, a cytokine with anti-angiogenic and immunostimulating properties, had a distinct effect on tumor growth [47]. Equally effective were also combinations of anti-TGFβ with agents stimulating immune response [48–50]. Finally, the combination of DNA vaccine directed against endoglin (CD105), a tumor vascular endothelial cell-surface protein, with interleukin-12 (IL-12) showed increased efficacy [51], although it had been shown that IL-12 alone is able to polarize the phenotype (M2 → M1) [52].
Related posts:
 Novel Strategy of Cancer Immunotherapy: Spiraling Up
Novel Strategy of Cancer Immunotherapy: Spiraling Up
 Dendritic Cell Vaccines for Cancer Therapy: Fundamentals and Clinical Trials
Psychoneuroendocrinoimmunotherapy of Cancer
Dendritic Cell Vaccines for Cancer Therapy: Fundamentals and Clinical Trials
Psychoneuroendocrinoimmunotherapy of Cancer
 Toll-Like Receptor Pathway and its Targeting in Treatment of Cancers
Toll-Like Receptor Pathway and its Targeting in Treatment of Cancers
 Photodynamic Therapy and Antitumor Immune Response
Photodynamic Therapy and Antitumor Immune Response
 Role of γδ T Lymphocytes in Cancer Immunosurveillance and Immunotherapy
Role of γδ T Lymphocytes in Cancer Immunosurveillance and Immunotherapy
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


