SUMMARY
The approximately 1 trillion platelets that circulate in an adult human are small anucleate cell fragments adapted to adhere to damaged blood vessels, to aggregate with one another, and to facilitate the generation of thrombin. These actions contribute to hemostasis by producing a platelet plug and then reinforcing plug strength by the action of thrombin converting fibrinogen to fibrin strands. To accomplish these tasks, platelets have surface receptors that can bind adhesive glycoproteins; these include the GPIb/IX/V complex, which supports platelet adhesion by binding von Willebrand factor, especially under conditions of high shear, and the αIIbβ3 (GPIIb/IIIa) receptor, which is platelet-specific and mediates platelet aggregation by binding fibrinogen and/or von Willebrand factor. Other receptors for adhesive glycoproteins (integrin α2β1 [GPIa/IIa], GPVI, and perhaps others for collagen; integrin α5β1 [GPIc*/IIa] for fibronectin; integrin α6β1 [GPIc/IIa] for laminin; and CLEC-2 for podoplanin) also contribute to platelet adhesion, but their precise contributions are less well defined. Activated platelets express both surface P-selectin, which mediates interactions with leukocytes, and CD40 ligand, which activates a number of proinflammatory cells, and release chemokines and a soluble form of CD40 ligand, thus initiating an inflammatory reaction. Platelet coagulant activity results from the exposure of negatively charged phospholipids on the surface of platelets and the generation of platelet microparticles, along with release and activation of platelet factor V and perhaps exposure of specific receptors for activated coagulation factor. Platelets change shape with activation as a result of a complex reorganization of the platelet membrane skeleton and cytoskeleton. With activation, platelets undergo release of α granules, dense bodies, and lysosomes, the contents of which work to restore vascular integrity. The activation process involves a number of receptors for agonists such as adenosine diphosphate, epinephrine, thrombin, collagen, thromboxane (TX) A2, vasopressin, serotonin, platelet activating factor, lysophosphatidic acid, sphingosine-1-phosphate, and thrombospondin, as well as several signal transduction pathways, including phosphoinositide metabolism, arachidonic acid release and conversion into TXA2, and phosphorylation of a number of different target proteins. Increases in intracellular calcium result from, and further contribute to, platelet activation. Platelet activation results in a change in the conformation of the integrin αIIbβ3 receptor, leading to high-affinity ligand binding and platelet aggregation.
Platelets also act as storehouses for a variety of molecules that affect platelet function, inflammation, innate immunity, cell proliferation, vascular tone, fibrinolysis, and wound healing; these agents are actively released upon platelet activation. Other vasoactive and platelet-activating substances are newly synthesized when platelets are activated. Through cooperative biochemical interactions, platelets can communicate with, and are affected by, other blood cells and endothelial cells.
Quantitative and qualitative disorders of platelets produce hemorrhagic diatheses (Chaps. 9 to 12). In pathologic states, uncontrolled platelet thrombus formation can lead to vasoocclusion and ischemic tissue necrosis, as, for example, in myocardial infarction and stroke (Chap. 25). Platelets may also facilitate tumor cell growth and metastasis.
Acronyms and Abbreviations:
AA, arachidonic acid; ADAM, a disintegrin and metalloprotease; ADMIDAS, adjacent to metal ion-dependent adhesion site; AngII, angiotensin II; APP, amyloid precursor protein; AP3, activator protein 3; BTK, Bruton tyrosine kinase; CIB, calcium and integrin binding protein; CLEC, C-type lectin-like receptor; COX, cyclooxygenase; DAG, diacylglycerol; DTS, dense tubular system; EDTA, ethylenediaminetetraacetic acid; EGF, epidermal growth factor; EMMPRIN, matrix metalloproteinase inducer; ERK, extracellular signal-regulated kinase; FAK, focal adhesion kinase; FOG, friend of GATA; FERM, four point one, ezrin, radixin, and moesin; GAS, growth arrest-specific gene; GP, glycoprotein; GPCR, G-protein–coupled receptor; GPI, glycosylphosphatidylinositol; GSK, glycogen synthase kinase; HDL, high-density lipoprotein; HPETE, hydroxyeicosatetraenoic acid; hTRPC, human canonical transient receptor potential; ICAM, intercellular adhesion molecule; IL, interleukin; IP3, inositol-1,4,5-trisphosphate; ITAM, immunoreceptor tyrosine-based activation motif; ITIM, immunoreceptor tyrosine-based inhibitory motif; ITSM, immunoreceptor tyrosine-based switch motif; JAM, junctional adhesion molecule; LAMP, lysosome-associated membrane protein; LDL, low-density lipoprotein; LIBS, ligand-induced binding site; LIMBS, ligand-associated metal binding site; LOX, lipoxygenase; LPA, lysophosphatidic acid; LPC, lysophosphatidyl choline; LPS, lipopolysaccharide; LT, leukotriene; LX, lipoxin; MAPK, mitogen-activated protein kinase; MIDAS, metal ion-dependent adhesion site; miRNA, microRNA; MLC, myosin light chain; MMP, matrix metalloproteinase; MRP, myeloid-related protein; MVB, multivesicular body; NAP, neutrophil-activating peptide; NET, neutrophil extracellular trap; NMR, nuclear magnetic resonance; NO, nitric oxide; PAF, platelet-activating factor; PAR, protease-activated receptor; PDGF, platelet-derived growth factor; PDI, protein disulfide isomerase; PDZ, postsynaptic density protein (PSD95), Drosophila disk large tumor suppressor (Dlg1), and zonula occludens-1 protein (zo-1); PECAM, platelet-endothelial cell adhesion molecule; PG, prostaglandin; PH, pleckstrin homology; PI, phosphoinositol; PIP2, phosphoinositol 4,5-bisphosphate; PIPK, phosphoinositol phosphate kinase; PKC, protein kinase C; PL, phospholipase; PNH, paroxysmal nocturnal hemoglobinuria; PPAR, peroxisome proliferator-activated receptors; PSGL, P-selectin glycoprotein ligand; PTB, phosphotyrosine binding; RIAM, Rap1GTP-interacting adapter molecule; SERT, serotonin transporter; SNP, single nucleotide polymorphism; S1P, sphingosine-1-phosphate; SR, scavenger receptor; STIM, stromal interaction molecule; SyMBS, synergy metal binding site; TFPI, tissue factor pathway inhibitor; TGF, transforming growth factor; TLR, toll-like receptor; TLT, TREM-like transcript; TNF, tumor necrosis factor; TP, thromboxane prostanoid receptor; TRAIL, TNF-related apoptosis-inducing ligand; TREM, triggering receptors expressed on myeloid cells; TSP, thrombospondin; TX, thromboxane; VASP, vasodilator-stimulated protein; VEGF, vascular endothelial growth factor; VWF, von Willebrand factor; WASP, Wiskott-Aldrich syndrome protein.
The hemostatic system is under elaborate control mechanisms lest the response be either inadequate to meet the hemorrhagic challenge or result in inappropriate thrombosis in response to trivial provocation. Evolutionary pressures have probably favored a more active hemostatic system as individuals with more active hemostatic systems were more likely to avoid death from hemorrhage prior to attaining sexual maturity or in association with childbirth. Our active hemostatic system may be less well adapted to our modern age, which is characterized by long life spans and progressive vascular disease, given that the deposition of a platelet-fibrin thrombus on a damaged atherosclerotic plaque is the cause of most myocardial infarctions and many strokes.
The platelet’s major function is to seal openings in the vascular tree. It is appropriate, therefore, that the initiating signal for platelet deposition and activation is exposure of underlying portions of the blood vessel wall that are normally concealed from circulating platelets by an intact endothelial lining (Fig. 2–1).1 Additional parameters that probably control the platelet response are: (1) the depth of injury, with deeper damage exposing more platelet-reactive materials and tissue factor (Chap. 5); (2) the vascular bed, with the blood vessels serving mucocutaneous tissues especially dependent on platelets for hemostasis, in contrast to the vascular beds in muscles and joints, which rely more on the coagulation mechanism; (3) the age of the individual, because the composition of the blood vessel wall probably changes with age; (4) the hematocrit, because increased numbers of erythrocytes enhance platelet interactions with the blood vessel wall by forcing platelets to the periphery of the bloodstream (as the erythrocytes disproportionately occupy the axial region), by imparting radially directed energy to platelets as the erythrocytes engage in flip-flop motions, and perhaps by releasing the platelet activator adenosine diphosphate (ADP) at sites of vascular injury2–4; and (5) the speed of blood flow and the size of the blood vessel, which will determine the number of platelets passing by a single point in a given time interval, the amount of time a platelet has to interact with the blood vessel wall or other platelets, the rate of dilution of platelet activating agents, and the forces tending to pull a platelet from the vessel wall or another platelet (shear rate).2,4–6 The vasospastic response that accompanies vascular injury, to which platelets contribute by release of thromboxane (TX) A2 and serotonin, probably plays a key role in decreasing hemorrhage and facilitating platelet and fibrin deposition via its effect on blood flow.
Figure 2–1.
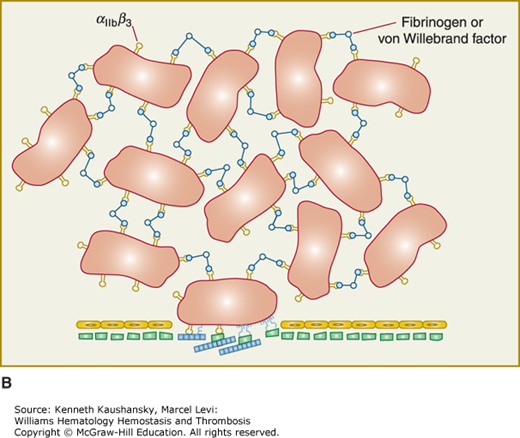
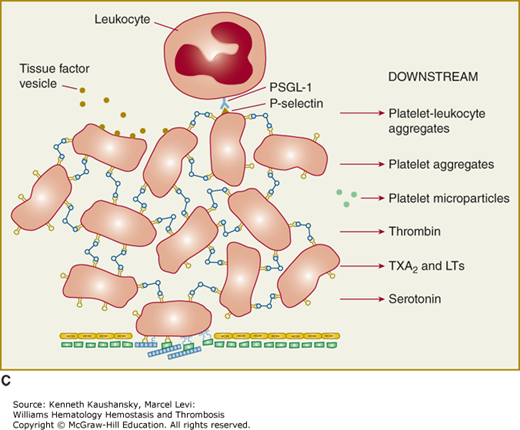
Platelet adhesion, activation, aggregation, and platelet–leukocyte interactions. A. Endothelial cells limit platelet deposition because they separate platelets from the adhesive proteins in the subendothelial area, produce two inhibitors of platelet function (nitric oxide [NO] and prostacyclin [PGI2]), and contain a potent enzyme (CD39) that can digest adenosine diphosphate (ADP) released from platelets. Platelet adhesion is initiated by loss of endothelial cells (or, in the case of an atherosclerotic lesion, rupture or erosion of the plaque), which exposes adhesive glycoproteins such as collagen and von Willebrand factor (VWF) in the subendothelium. In addition, VWF and perhaps other adhesive glycoproteins in plasma deposit in the damaged area, in part by binding to collagen. Platelets adhere to the subendothelium via receptors that bind to the adhesive glycoproteins. Glycoprotein (GP) Ib binding to VWF plays a prominent role, but integrin α2β1 (GPIa/IIa) and GPVI binding to collagen and other platelet receptors (see Table 2–4) probably also play a role. After platelets adhere, they undergo an activation process that leads to a conformational change in integrin αIIbβ3 receptors involving headpiece extension and leg separation (see Fig. 2–5), resulting in their ability to bind with high-affinity select multivalent adhesive proteins, most prominently fibrinogen and VWF, including the VWF that binds to collagen in the subendothelial area. B. Platelet aggregation occurs when the multivalent adhesive glycoproteins bind simultaneously to integrin αIIbβ3 receptors on two different platelets, resulting in receptor crosslinking. Clustering of the receptors probably also contributes to the stability of the aggregates (not shown). C. After platelets adhere and aggregate, they help to initiate coagulation by binding tissue factor-containing vesicles circulating in the plasma, exposing negatively charged phospholipids on their surface (not shown), releasing platelet factor V (not shown), and releasing procoagulant microparticles. Activated platelets also express P-selectin on their surface, which leads to recruitment of leukocytes via interactions between platelet P-selectin and P-selectin glycoprotein ligand-1 (PSGL-1) expressed on the surface of leukocytes. Other interactions between platelets and leukocytes are detailed in Fig. 2–9. Thrombus formation is a dynamic cyclical process, with platelets repeatedly adhering, aggregating, and then breaking off and embolizing downstream. Platelet–leukocyte aggregates, platelet aggregates, platelet microparticles, thrombin, thromboxane A2 (TXA2), leukotrienes (LTs), and serotonin probably all go downstream and affect the microvasculature. Ultimately, the vessel either becomes fully occluded or loses its thrombogenic reactivity; that is, it becomes passivated.
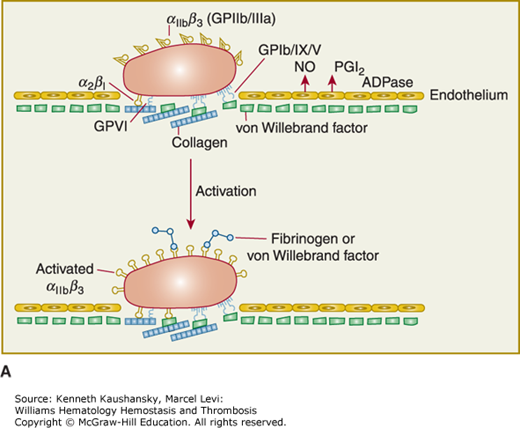
The initial adhesion of platelets occurs to the adhesive proteins within the subendothelial layer immediately subjacent to the endothelium1,5 or to activated endothelium. The platelet expresses many receptors that participate in adhesive interactions (Table 2–1). Intravital microscopy and ex vivo flow chamber studies indicate that discoid platelets that show minimal or no evidence of activation can form the initial layers of platelet aggregates when laminar flow is disrupted by a stenotic lesion, but that stable thrombus development requires the generation and/or release of soluble activators.6 Membrane tethers, which can undergo restructuring and stabilization, are important in achieving interactions with matrix proteins and other platelets.
| Protein | Properties |
|---|---|
| Actin1805 | Mr = 42,000 20–30% of total platelet protein (0.55 M; 2 × 106 per platelet) β and γ forms present at a ratio of 5:1 Monomeric actin (G-actin) bound to calcium-ATP (or adenosine diphosphate [ADP]) Polymerization requires energy (ATP→ADP) and produces F-actin F-actin filaments: two strands of intertwined helices with polarity based on ability to interact with myosin fragment (“pointed” and “barbed” ends) Steady-state polymerization: monomers lost from pointed end while others join barbed end (“treadmilling”) |
| Profilin1806 | Mr = 15,200 Forms 1:1 reversible complex with actin monomer Prevents actin polymerization May help “recharge” actin monomers with ATP |
| Gelsolin1807 | Mr = 81,000 (5 μM; 2 × 104 per platelet) Binds to barbed end of F-actin filaments Severs actin filaments Facilitates nucleation Produces shorter filaments with gel→sol transformation |
| Thymosin β4267,268 | Mr = 5000 (0.55 M; 2 × 106 per platelet) Binds actin monomer Inhibits actin polymerization |
| Tropomyosin1808 | Mr = 28,000; rod-shaped dimer of 35-nm length Binds to groove on actin filaments (6 actins:1 tropomyosin) Not all actin filaments have bound tropomyosin |
| Caldesmon1809 | Mr = 80,000; asymmetric Binds to actin, tropomyosin, myosin, and calmodulin May control actin filament bundling and actomyosin adenosine triphosphatase (ATPase) |
| Filamin A (X) and B (3) (actin-binding protein)133,154,216,249,1810,1811 | Filamin A-to-B = 10:1 Mr = 260,000 subunit; tail-to-tail dimer; elongated 162-nm flexible rod composed of 24 immunoglobulin-like domains; phosphorylated 2–3% of platelet protein Binds actin with 1 actin binding protein molecule per 14 actin molecules Binds glycoprotein (GP) Ibα and integrin β subunit cytoplasmic domains and links GPIb/IX to actin Binds small guanosine triphosphatases (GTPases) ralA, ras, rho, Cdc-42, as well as kinases and phosphatases, and exchange factors Trio and Toll Crosslinks actin filaments to form a gel Dephosphorylation leads to loss of activity |
| Migfilin142,1812 | Mr = 50,000; binds kindling-2 and vasodilator-stimulated protein (VASP) Can displace filamin from β3 cytoplasmic domain, facilitating binding of talin |
| Talin142,245,1812,1814 | Mr = 235,000 3% of platelet protein Binds to β3 integrin cytoplasmic tail to activate αIIbβ3; also binds vinculin and α-actinin; cleaved and activated by calpain |
| α-Actinin1806 | Mr = 100,000 and 102,000; dimer Binds actin at 1:10 stoichiometry; binds Ca2+ Forms gel with F-actin; cooperates with actin-binding protein; promotes actin polymerization |
| Vinculin269,1815,1816 | Mr = 130,000 Binds to talin; may link actin to membrane proteins at adhesion sites |
| Myosin II1817,1818 | Mr = 480,000 (2 × 200,000; 2 × 20,000; 2 × 16,000) 2–5% of platelet protein; 325 × 111-nm filaments Myosin light chain (Mr = 20,000); phosphorylated; required for ATPase activity |
| Myosin light-chain kinase1819 | Mr = 105,000 Phosphorylates myosin light chain and activates actomyosin ATPase leading to contraction |
| Calmodulin1820 | Mr = 17,000 Binds four calciums and activates myosin light-chain kinase |
| CapZ154,216 | Mr = 36,000 and 32,000 (5 μM; 2 × 104 per platelet) Heterodimer Binds barbed ends of actin filaments |
| Cofilin154,216 | Mr = 20,000 Accelerates depolymerization of actin filaments |
| Fimbrin (L-plastin) | Mr = 68,000 Bundles actin filaments Found in microvilli |
| VASP154,216 | Mr = 50,000 Tetrameric Binds profilin, vinculin, zyxin |
| GTPases154,229,249 | Cdc42–filopodia Rho–stress fibers Rac–lamellipods and ruffles Rap1b–αIIbβ3 control |
| Tyrosine kinases | pp60src pp125Fak–αIIbβ3 signaling pp72syk–GPVI signaling |
| Adaptor proteins | 14–3-3ζ–binds to GPIbα Pleckstrin–phosphorylated on activation |
| PI kinases | PI-3 kinase PI4P-5 kinase |
| Spectrin | α,β heterodimers form head to head tetramers Bind to actin filaments |
| α,γ Adducins | Cap barbed ends of actin filaments and bind to spectrin Phosphorylated with platelet activation and cleaved by calpain |
The shear rate differentially affects platelet adhesion to surfaces.3,4,7–12 Shear rates, which reflect the differences in flow velocity as a function of distance from the blood vessel wall, vary considerably throughout the vasculature, being highest in small arterioles and lowest in large arteries and veins; very high rates are observed at the tips of severely stenotic atherosclerotic arteries.6,11,12 Very high shear rates can cause platelets to aggregate via a mechanism that involves von Willebrand factor (VWF) binding to glycoprotein (GP) Ib/IX followed by intracellular signaling, leading to activation of integrin αIIbβ3.13–16 Platelets contribute more significantly to arterial thrombi than to venous thrombi, perhaps as a result of differences in the shear rates in the different beds.5
Platelets also interact directly with exposed collagen, including types I, III, and VI, via GPVI and integrin α2β1 (GPIa/IIa), or perhaps one or more of the many other receptors implicated in platelet–collagen interactions (e.g., CD36 [GPIV], p65).17–29 The interaction of platelets with collagen is most evident at relatively low shear rates. Depending on the vascular bed, available adhesive glycoproteins, and shear conditions, it is likely that various combinations of platelet receptors, including GPIbα, integrin α2β1 (GPIa/IIa), GPVI, and integrin αIIbβ3, act in concert to transform the tethering and slow translocation of platelets initiated by GPIbα interacting with VWF into stable platelet adhesion.1,3,4,8,10,16,25,28
For platelet plug formation to occur, platelets must undergo activation as well as adhesion. Adhesion of platelets to subendothelial structures, in particular VWF at high shear, may itself lead to platelet activation, including the generation of TXA2, release of ADP and serotonin, and activation of the integrin αIIbβ3 receptors on the luminal side of the platelet so that they adopt their high-affinity ligand-binding conformation(s).10 These positive feedback mechanisms ensure an adequate hemostatic response. Depending on the nature of the surface to which they adhere, platelets also undergo variable spreading reactions and become anchored by a process that at least partially involves integrin αIIbβ3 ligation and clustering, leading to “outside-in” signaling, cytoskeletal reorganization, and tyrosine phosphorylation; these reactions also contribute to initiating the release reaction.30–36 In addition, platelet activators, such as ADP, are released or synthesized at the site of vascular injury, resulting in a local response. Cooperative biochemical interactions between erythrocytes and platelets may enhance platelet activation.37
Activated luminal integrin αIIbβ3 receptors on adherent platelets bind VWF, fibrinogen, and other adhesive glycoproteins, and await the interaction with another platelet, which itself may have undergone activation of its integrin αIIbβ3 receptors as a result of exposure to released ADP and TXA2. Alternatively, a platelet may become activated and bind VWF or fibrinogen while still circulating, in which case the platelet-ligand complex may bind directly to an activated integrin αIIbβ3 receptor on the luminal surface. The binding of adhesive ligands to platelet receptors then repeats itself, resulting in the recruitment of additional layers of platelets, and ultimately the formation of a hemostatic plug. Intravital videomicroscopy of the mesenteric and cremasteric circulations of mice after endothelial cell damage demonstrates that, at least in these vascular beds, platelet thrombus formation is initially a very dynamic process, with many platelets depositing but then embolizing.38 The thrombus grows relatively slowly compared to what its growth would be if all of the platelets that deposited remained attached to the surface.39–41
The integrin αIIbβ3 receptor occupies a central role in determining the extent of platelet aggregation, in part because it is present at an extraordinarily high density on the platelet surface (approximately 50,000 receptors per platelet, such that receptors are probably less than 20 nm apart).30,42–45 This permits it to rapidly initiate platelet aggregation. On the other hand, the receptor is not in its high-affinity ligand-binding state on resting platelets but rather needs to be activated by agonists, including ADP, serotonin, thrombin, collagen, and TXA2, that are localized to sites of vascular injury.34,44,46 As a result, platelets can circulate in plasma containing high concentrations of the integrin αIIbβ3 ligands fibrinogen and VWF without ongoing platelet thrombus formation. The agonists that activate the integrin αIIbβ3 receptor are likely to work in combination in vivo. In fact, the mixture of agonists present is likely to change as the process unfolds, with collagen perhaps more important at the beginning, thrombin more important later on, and the other agonists in varying mixtures throughout. The platelet activation effects of multiple agonists may be additive or synergistic, depending on the mechanism(s) involved.47,48
A number of mechanisms stabilize platelet aggregates. These include absence of fibrinogen (presumably limiting fibrin formation),41 leptin,49–51 CD40 ligand,52 growth arrest-specific gene 6 product (Gas6) and its receptors (Axl, Sky, and Mer),53–57 Eph kinases and ephrins,58 factor XII,59 plasminogen activator inhibitor-1 and vitronectin,50 or inhibition of select regions of fibrinogen.60
Activated platelets can facilitate thrombin generation by one or more different mechanisms, including recruitment of bloodborne tissue factor, synthesis or activation of tissue factor, formation of procoagulant microvesicles, exposure of activated factor V, exposure of negatively charged phospholipids, and perhaps activation of the contact system. The thrombin thus generated further activates platelets, leading to more extensive degranulation; it also further activates coagulation and initiates the deposition of fibrin strands that reinforce the platelet thrombus and serve as sites for additional VWF deposition.61 Thrombin also helps to consolidate the plug by initiating platelet-mediated clot retraction (see section “Platelet Shape Change, Spreading, Contraction, and Clot Retraction” below). Finally, thrombin affects the surface membrane receptors, downregulating GPIb/IX and upregulating integrin αIIbβ3, perhaps facilitating the transition from platelet adhesion to platelet aggregation.62–65
Release of vasoactive and mitogenic agents, as well as chemokines, from platelets contributes to the inflammatory response, as does the appearance of P-selectin on the surface of activated platelets and endothelial cells, because P-selectin and other platelet receptors recruit leukocytes to the damaged region.66–68 Finally, after contributing to hemostasis and initiating an inflammatory response, platelet-fibrin thrombi eventually resolve, most likely by a combination of embolization, fibrinolysis, and macrophage removal of debris.
Several inhibitory factors serve to balance platelet activation and thus prevent excessive platelet deposition. The dilutional effects of flowing blood are probably most important; thus, alterations in the surface of the blood vessel that produce local areas of stasis in which platelets and coagulation factors may concentrate are prothrombogenic.2,5 Endothelial cells can synthesize two potent inhibitors of platelet activation, prostacyclin and nitric oxide (Chap. 5).69–72 Generation of prostacyclin at sites of vascular injury or inflammation may provide a mechanism to limit platelet accumulation. Nitric oxide, which is synthesized by endothelial cells, is a potent inhibitor of ex vivo platelet adhesion and aggregation. Endothelial cells and lymphocytes also have CD39, an ecto-ATP diphosphohydrolase (ecto-ADPase) that can digest ATP and ADP to adenosine monophosphate (AMP), and thus limit the effects of released ADP.73,74 They also have CD73, which can convert AMP into the platelet inhibitor adenosine.
On films made from blood anticoagulated with the strong calcium chelating agent ethylenediaminetetraacetic acid (EDTA) and treated with Wright stain, platelets appear as small bluish-gray, oval-to-round–shaped cell fragments with several purple-red granules. The mean diameter of platelets varies in different individuals, ranging from approximately 1.5 to 3.0 μm, approximately one-third to one-fourth that of erythrocytes. There is also considerable variability in the size of platelets in a single individual, with occasional platelets in normal blood samples having diameters greater than half the diameter of erythrocytes. Overall, platelet size appears to follow a log normal distribution with an average volume of approximately 7 fL.75 When unanticoagulated blood is used to prepare blood films, platelets undergo variable activation and spreading, and thus platelet aggregates are commonly seen; platelets from such specimens may demonstrate three or four very long finger-like processes extending out from the body of the platelet (filopodia), and some platelets may be devoid of granules.
Electron microscopy reveals a fuzzy coat (glycocalix) extending 14 to 20 nm from the platelet surface, which is thought to be composed of membrane GPs, glycolipids, mucopolysaccharides, and adsorbed plasma proteins (Fig. 2–2).76 Platelets move in an electric field as if they have a net negative surface charge; sialic acid residues attached to proteins and lipids are major contributors to this negative charge.77 The electrostatic repulsion created by the negative surface charge may help prevent resting platelets from attaching to each other or to negatively charged endothelial cells.
Figure 2–2.
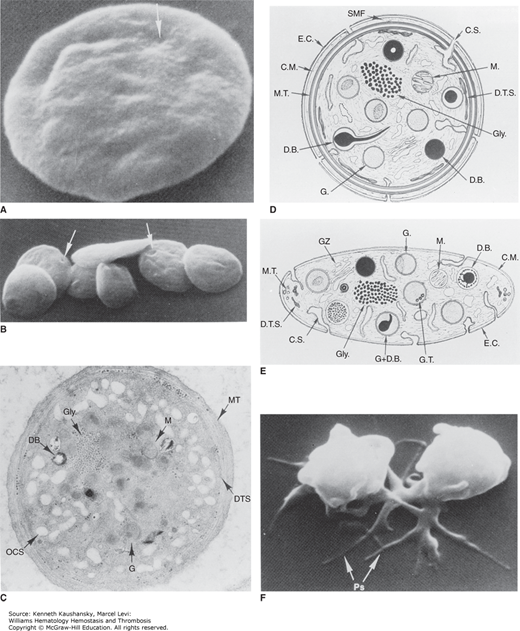
A and B. Discoid platelets. The lentiform shape of blood platelets is well preserved in samples fixed in glutaraldehyde and critical point dried for study in the scanning electron microscope. The indentations apparent on the otherwise smooth surfaces of the platelets (arrows) indicate sites where channels of the open canalicular system (OCS) communicate with the cell exterior. (Magnification: A, ×13,200; B, ×35,000.) C, D, and E. Ultrastructural features observed in thin sections of discoid platelets cut in the equatorial plane (C and D) or cross-section (E). Components include the exterior coat (E.C.), trilaminar unit membrane (C.M.), and submembrane area containing the specialized filaments of the membrane skeleton (SMF). The plasma membrane indentations form the walls of the channels of the surface-connected open canalicular system (C.S. and OCS). The circumferential band of microtubules (M.T.) is seen as a continuous band beneath the plasma membrane on the equatorial section and as small open cylinders at the ends of the platelet on the cross-section. Glycogen granules (Gly.) are prominent punctate structures in the cytoplasm, and residual Golgi zones (GZ) can also be identified. Organelles include mitochondria (M.), dense bodies (D.B.), and α granules (G.), many of which have regions of electron density (nucleoids). The dense tubular system (D.T.S.), the platelet equivalent of the sarcoplasmic reticulum, sequesters calcium. (Magnification: C, ×30,000.) F. Platelet shape change. Platelets exposed to adenosine diphosphate and then fixed and examined by scanning electron microscopy. The platelets lose their discoid shape and become spiny spheres with long extensions, variably referred to as filopodia or pseudopodia. (Magnification: ×17,000.) (Reproduced with permission from Bloom AL, et al: Hemostasis and Thrombosis. Edinburgh: Churchill Livingstone; 1994.)
Indentations on the platelet surface are thought to be the openings of the open canalicular system, which is an elaborate channel system composed of invaginations of the plasma membrane that extend throughout the platelet (see Fig. 2–2 and “Membrane Systems” below). The contents of platelet granules can gain access to the outside when the granules fuse with either the plasma membrane or any region of the open canalicular system. Similarly, glycoproteins contained within granule membranes can join the plasma membrane after granule fusion with either the plasma membrane or the open canalicular system.
The plasma membrane is a trilaminar unit composed of a bilayer of phospholipids embedded with cholesterol, glycolipids, and glycoproteins.76,78 Platelets prepared by the freeze–fracture technique demonstrate more intramembranous particles embedded in the outer platelet membrane leaflet than in the inner leaflet, which is the reverse of findings in erythrocytes; this observation presumably reflects the many external receptors that mediate platelet interactions. The plasma membrane is thought to contain the sodium- and calcium-adenosine triphosphatase (ATPase) pumps that control the intracellular ionic environment of the platelet. Approximately 60 percent of platelet phospholipids are contained in the plasma membrane. The phospholipids are asymmetrically organized in the plasma membrane; the negatively charged phospholipids are almost exclusively present in the inner leaflet, whereas the others are more evenly distributed.79 The negatively charged phospholipids, especially phosphatidylserine, are able to accelerate several steps in the coagulation sequence, and so their presence in the inner leaflet of resting platelets, separated from the plasma coagulation factors, is thought to be a control mechanism for preventing inappropriate activation of the coagulation system.80,81 During platelet activation induced by select agonists, the aminophospholipids may become exposed on the platelet surface or on the surface of microparticles (see “Platelet Coagulant Activity” below).80–83
The phospholipid asymmetry in resting platelets may be maintained by an ATP-dependent aminophospholipid translocase that actively moves phosphatidylserine and phosphatidylethanolamine from the outer to the inner leaflet.80,84 Interactions of negatively charged phospholipids with cytoskeletal or other cytoplasmic elements may also contribute to the asymmetry.80,81,85,86
Lipid rafts are dynamic, cholesterol- and sphingolipid-rich membrane microdomains that are important in signaling and intracellular trafficking. In platelets, the cholesterol-to-phospholipid molar ratio is twofold higher in rafts than in bulk membranes, with sphingomyelin accounting for the majority of total raft lipids.87 Platelet lipid rafts contain the marker proteins flotillin 1, flotillin 2, stomatin, and the ganglioside GM1; the rafts are also notable for being devoid of caveolin. Other proteins, such as CD36, CD63, CD9, integrin αIIbβ3, and glucose transporter (GLUT)-3, are present in rafts prepared from resting platelets.87 Upon activation of GPVI, Fc gamma chain, FcγRIIa, and GPIb/IX/V partition into the lipid rafts,88,89 as do c-Src,90 phosphatidic acid, and phosphoinositol (PI) 3′-kinase (PI3K) products.87,91 Factor XI binds to extracellularly oriented lipid rafts and undergoes activation.92 The calcium entry channel hTRPc1 is associated with lipid rafts in platelets and, upon platelet activation, contributes to calcium entry that is regulated by the state of intracellular calcium stores (store-mediated calcium entry).93 The functionally detrimental effects of chilling platelets are thought to be mediated, at least in part, by the temperature-dependent coalescence of platelet lipid rafts.94
Open Canalicular System The surface-connected open canalicular system is an elaborate series of conduits that begin as indentations of the plasma membrane and tunnel throughout the interior of the platelet.76,95,96 Tracer studies demonstrate that the open canalicular system is contiguous with the exterior of the platelet, even though elements of the open canalicular system may appear as closed vesicles or vacuoles by electron microscopy of sectioned platelets.76,95–97
The open canalicular system may serve several functions. It provides a mechanism for entry of external elements into the interior of the platelet. It also provides a potential route for the release of granule contents to the outside, eliminating the need for granule fusion with the plasma membrane itself.97,98 This latter function is especially important because, under most circumstances, platelet granules appear to move to the center of the platelet upon platelet activation rather than to the periphery.76,95,99 Controversy remains, however, regarding the relative frequency with which secretion occurs via the open canalicular system versus direct fusion with the plasma membrane.76,95,100
The open canalicular system also represents an extensive internal store of membrane. Both filopodia formation and platelet spreading after adhesion require a dramatic increase in surface plasma membrane compared to the plasma membrane of resting platelets, and it is not possible for new membrane to be synthesized during the short time-course of these phenomena. Thus, the membrane of the open canalicular system most likely contributes to the increase in plasma membrane under these conditions; the membranes of α granules, dense bodies, and, to a lesser extent, lysosomes may also contribute, but only if the stimulus is sufficient to induce the fusion of these organelles with the plasma membrane (release reaction). Finally, the membrane of the open canalicular system may serve as a storage site for plasma membrane glycoproteins. For example, under certain conditions, platelet activation by thrombin leads to a consistent, selective loss of GPIb/IX from the platelet surface, and data from electron microscopy indicate that the GPIb/IX becomes sequestered in the open canalicular system.63,64,101 Plasmin may produce a similar phenomenon.101,102 Platelet activation leads to an increase in surface integrin αIIbβ3, and although much of this receptor is thought to derive from α-granule membranes, at least some may come from integrin αIIbβ3 in the membranes of dense bodies and the open canalicular system.101,103 Similarly, GPVI, the P2Y1 ADP receptor, and the TXA2 receptor, and perhaps other receptors, are present in the open canalicular system and can be recruited to the platelet surface with activation.104,105
Dense Tubular System/Sarcoplasmic Reticulum The dense tubular system (DTS) is a closed-channel network of residual endoplasmic reticulum characterized histocytochemically by the presence of peroxidase activity.76,106–108 The channels of the DTS are less extensive than those of the open canalicular system and tend to cluster in regions in close approximation to the open canalicular system.76 The DTS is analogous to the sarcoplasmic reticulum of muscle because it can sequester Ca2+ and release it when platelets are activated, leading to shape change, granule centralization, and secretion.109,110 Calreticulin, a calcium binding protein found in the DTS/sarcoplasmic reticulum, probably helps to sequester ionized calcium.111,112 Release of Ca2+ from the DTS/sarcoplasmic reticulum involves the binding of inositol-1,4,5-trisphosphate (IP3), a messenger molecule formed during signal transduction, to IP3 type II receptors on the DTS/sarcoplasmic reticulum membrane (Fig. 2–3).113,114 Cyclic AMP inhibits Ca2+ release from the DTS/sarcoplasmic reticulum, either by enhancing the calcium pumping mechanism115 or by inhibiting release induced by IP3.116 NO inhibits Ca2+ uptake by the DTS/sarcoplasmic reticulum at high concentrations and stimulates uptake at low concentrations by effects on the calcium ATPase(s) SERCA26 and SERCA3.117,118 Depletion of intracellular calcium stores activates store-operated calcium entry (SOCE) into platelets (reviewed in Ref. 119). The depletion of Ca2+ from the DTS/sarcoplasmic reticulum is sensed by stromal interaction molecule 1 (STIM1), a transmembrane protein with a Ca2+ binding motif (EF hand) in the DTS/sarcoplasmic reticulum.120–122 Loss of Ca2+ binding to STIM1 results in translocation and activation of Orai1, a calcium release activated calcium (CRAC) channel in the plasma membrane,123,124 that allows Ca2+ entry into the platelet. Although mice with defects in STIM1 and Orai1 have demonstrated abnormalities in platelet function,120–122 humans with mutations in these proteins have had immune dysfunction, but no overt hemostatic or thrombotic abnormalities.125–127 The human canonical transient receptor potential 1 (hTRPC1) has also been implicated in regulating platelet SOCE, but mice deficient in this protein do not have a defect in platelet Ca2+ entry.128–130
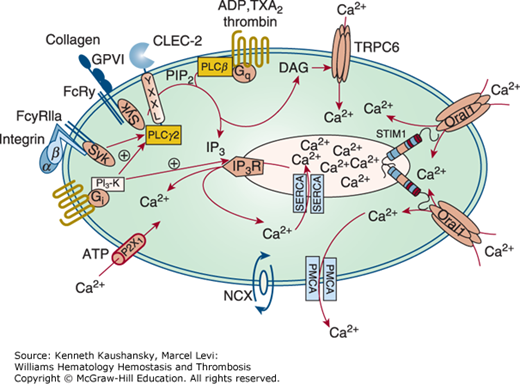
Figure 2–3.
Platelet calcium homeostasis. Upon receptor activation, different phospholipase (PL) C isoforms hydrolyze phosphatidylinositol-4,5-bisphosphate (PIP2) to inositol-1,4,5-trisphosphate (IP3) and diacylglycerol (DAG). IP3 releases Ca2+ from the intracellular stores in the dense tubular system (DTS)/sarcoplasmic reticulum. The transmembrane protein stromal interaction molecule 1 (STIM1) senses the reduction in Ca2+ through a decrease in Ca2+ occupancy of its EF hand domain and then opens Orai1 Ca2+ channels in the plasma membrane, a process called store-operated calcium entry, whereas DAG mediates calcium entry through canonical transient receptor potential channel 6 (TRPC6). Additionally, a direct receptor-operated calcium (ROC) channel, P2X1, and an Na+/Ca2+ exchanger (NCX) contribute to the elevation in Ca2+ in the platelet cytoplasm. The counteracting mechanisms to replenish DTS/sarcoplasmic reticulum Ca2+ stores involve Ca2+ adenosine triphosphatases (ATPases) (SERCAs). Plasma membrane Ca2+ ATPases (PMCAs) pump Ca2+ through the plasma membrane out of the cell. ADP, adenosine diphosphate; CLEC-2, C-type lectin-like receptor 2; FcRγ, Fc receptor γ chain; FcγRIIa, Fc γ receptor IIa; GPVI, glycoprotein VI; IP3R, IP3 receptor; PI3-K, phosphatidylinositol 3-kinase; Syk, spleen tyrosine kinase. Because of controversies about the localization and role of TRPC1 in the literature, this protein is not depicted in the figure. (Adapted with permission from Varga-Szabo D, Braun A, Nieswandt B: Calcium signaling in platelets, J Thromb Haemost. 2009 Jul;7(7):1057–1066.)
The DTS membrane is also probably a major site of prostaglandin and TX synthesis109,131; in fact, the peroxidase activity used to identify the DTS is an enzymatic component of prostaglandin synthesis.131,132
The discoid shape of the resting platelet is maintained by a well-defined and highly specialized cytoskeleton. This system of molecular struts and girders preserves the shape and integrity of the platelet as it encounters high shear forces in the circulation. The platelet cytoskeleton is operationally defined as proteins that are insoluble in the presence of the nonionic detergent Triton X-100 under defined ionic conditions. The three major cytoskeletal elements are the spectrin membrane skeleton, the marginal microtubule coil, and the actin cytoskeleton.
Membrane Skeleton The plasma membrane and open canalicular system of the resting platelet are supported by a highly structured cytoskeletal system (see Figs. 2–2 and 2–4). This two-dimensional network, located just beneath the plasma membrane, has remarkable structural resemblance to its red blood cell counterpart. Thus, both involve the self-assembly of elongated spectrin strands that interconnect through their binding to actin filaments, generating triangular pores. Platelets contain approximately 2000 molecules of spectrin.133–136 The spectrin network coats the cytoplasmic surfaces of both the plasma membrane and the open canalicular system. In contrast to the erythrocyte membrane skeleton, however, in which spectrin molecules connect on short actin filaments, in platelets, spectrin joins into a network by binding to the ends of actin filaments in close apposition to the plasma membrane. As a result, the spectrin lattice is assembled into a continuous network by its association with actin filaments. Moreover, tropomodulins, which are abundant in erythrocytes, are not expressed at significant levels in platelets and thus are unlikely to play a role in capping the pointed ends of actin filaments. Instead, these ends appear to be free in resting platelets. Finally, the protein adducin is abundantly expressed in platelets and appears to cap the majority of the barbed ends of the filaments making up the resting platelet cytoskeleton.137 This serves to target them to the spectrin-based membrane skeleton, as the affinity of spectrin for adducin-actin complexes is greater than for either adducin or actin alone.138–140
Figure 2–4.
Diagrammatic depiction of established and hypothetical connections between select platelet transmembrane glycoproteins and the underlying membrane skeleton. Although evidence exists for direct interactions between IIb 3 with talin and Src and between GPIb with 14–3–3 and filamin, the remainder of the interactions are only hypothetical and are based on the recovery of proteins in the membrane skeleton fraction of solubilized platelets. (Adapted with permission from Colman RW: Hemostasis and Thrombosis: Basic Principles and Clinical Practice. 4th edition. Philadelphia, PA: Williams & Wilkins; 2001.)
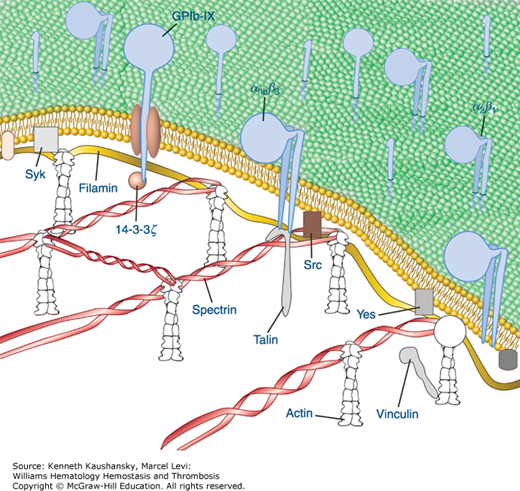
The platelet spectrin-actin filament network is fortified by interactions with filamin A (actin binding protein), a noncovalent dimer of two identical Mr 280,000 protein subunits that fastens GPIb/IX/V complexes to the sides of actin filaments. By interacting with both the transmembrane glycoprotein GPIbα and the actin immediately below the membrane, filamin A connects these components to the spectrin network and the resulting membrane cytoskeleton, probably contributing to the platelet’s discoid shape. In addition, the association of GPIbα with the membrane skeleton restricts the expansion of the spectrin network and probably helps to organize receptors into linear arrays on the platelet surface, thus enhancing receptor cooperation (see Fig. 2–4).133 Filamin also binds to the cytoplasmic domains of the β3 subunits of integrin receptors, and this keeps the receptor in a low-affinity state.141–143 Other proteins that have been found in the membrane skeleton include talin, vinculin, dystrophin-related protein, molecules implicated in signal transduction, and several isoenzymes of protein kinase C.133
Talin has been implicated in controlling integrin αIIbβ3 activation, by binding to the cytoplasmic domain of integrin β3 when phosphorylated and/or cleaved by calpain (see “Integrin αIIbβ3” below and Fig. 2–4).144–148 Migfilin (filamin-binding LIM protein-1) is a 373-amino-acid protein of Mr 50,000 that can displace filamin from the integrin β3 cytoplasmic domain, thus facilitating talin binding and activation. Moreover, joining integrin αIIbβ3 to the membrane skeleton via an integrin β3 linkage creates the possibility for an actin–myosin contraction process to supply sufficient force to integrin αIIbβ3 to induce conformational changes in the receptor that result in high-affinity ligand binding.149 The protein vimentin (Mr 58,000), which is an important component of intermediate filaments, is present in platelets and contributes to the membrane cytoskeleton. When platelets are activated, vitronectin–plasminogen activator inhibitor-1 (PAI-1) complexes bind to surface vimentin where they are strategically located to inhibit fibrinolysis.150 With platelet activation, integrins αIIbβ3 and α2β1 join the cytoskeleton. Thus, the cytoskeleton may affect whether receptors are free to move in the plane of the membrane; it may also have a role in moving certain receptors from the surface to the interior of platelets and vice versa via the open canalicular system.101,133 The membrane skeleton may also be important in platelet spreading after adhesion.
Microtubules One of the most distinguishing features of the resting platelet is its marginal microtubule coil (see Fig. 2–2). Located below the plasma membrane, it plays an important role in platelet formation from megakaryocytes and maintaining the platelet’s discoid shape.76,151–153 Microtubules are the largest cytoskeletal filaments (25 nm) and are comprised of hollow polarized polymers composed of 13 protofilaments made up of αβ tubulin dimers (each of Mr 110,000) that associate with several high-molecular-weight proteins (microtubule-associated proteins).153–155 Motor proteins of the dynein and kinesin families are also associated with microtubules.156–158 In cells, αβ tubulin subunits are in dynamic equilibrium with assembled microtubules such that reversible cycles of assembly and disassembly of microtubules are frequently observed.159 The critical concentration for tubulin polymerization is 5 μM, which is well below the tubulin concentration in platelets (70 μM), and thus, 60 percent of platelet tubulin is present as polymer.154,160 On cross-section, approximately eight to 12 separate hollow structures are observed at the tapered ends of the platelet (see Fig. 2–2). Direct visualization of microtubule assembly in resting mouse platelets indicates that the circumferential coil in platelets is composed of at least eight actively polymerizing microtubules.159 Microtubule dynamics allow for necessary changes in platelet shape that occur during the platelet life span and with activation. Tubulin is acetylated in resting platelets and undergoes deacetylation by histone deacetylase (HDAC) 6 with activation in association with the dissolution of the marginal band.161,162
Platelets contain four different tubulin isoforms (β1, β2, β4, β5), but β1 is dominant and is specific for megakaryocytes and platelets. Targeted gene deletion of β1-tubulin in mice results in thrombocytopenia and abnormal platelet and microtubule morphology.153 β1-Tubulin–deficient platelets are spherical in shape, probably as a result of having defective marginal bands with fewer (approximately two to three) than normal (approximately eight) microtubule coils.163 A heterozygous polymorphism of human β1-tubulin (Q43P) has been described in association with macrothrombocytopenia,164 but it is probably not causal,165 and individuals homozygous for the Q43P variant have low platelet counts, abnormal platelet ultrastructure, and decreased tubulin, but normal platelet length, width, and area.166 A heterozygous β1-tubulin mutation (R207H) in a strategically located region of the molecule has been reported in association with macrothrombocytopenia, as has an F260S mutation167 and an R318W mutation165 (Chap. 10).168
Actin Filaments Actin is the most abundant of all platelet proteins, with 2 million molecules expressed per platelet (0.5 mM).169 Like tubulin, actin is in dynamic monomer-polymer equilibrium, with 40 percent of the actin subunits polymerized to form 2000 to 5000 linear actin filaments in resting platelets (Fig. 2–5). The rest of the actin in the platelet cytoplasm is maintained in storage as a 1:1 complex with β4-thymosin; this stored actin is converted to filaments during platelet activation to drive cell spreading.170 Thus, actin filaments crisscross the interior of the cell, interconnected at various points into a rigid cytoplasmic network by abundantly expressed actin crosslinking proteins, including filamin and α-actinin.171–173 Filamin exists in solution as homodimers of subunits that themselves are elongated strands composed primarily of 24 repeats, each approximately 100 amino acids in length, that are folded into immunoglobulin (Ig) G-like β barrels.174,175 There are three filamin genes, and they are located on the X chromosome, chromosome 3, and chromosome 7.176,177 Filamin A and filamin B are expressed in platelets, with filamin A accounting for approximately 90 percent of total filamin.
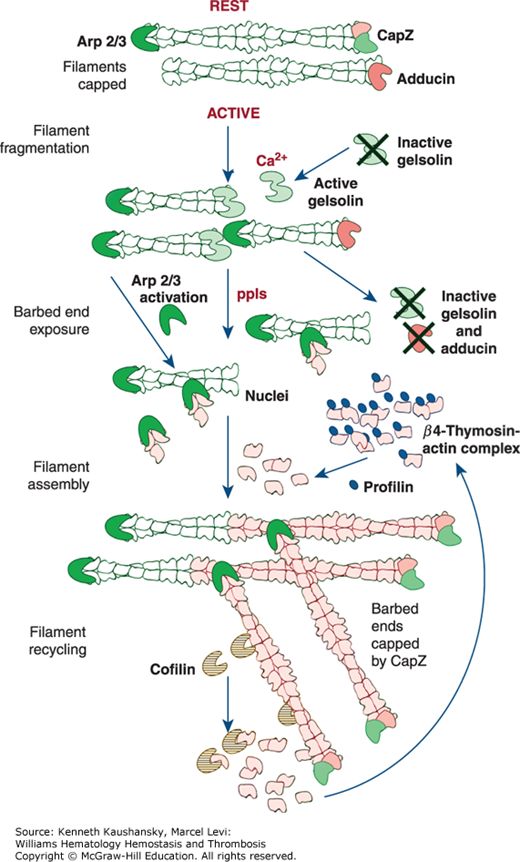
Figure 2–5.
Control of platelet actin assembly. (Rest) Forty percent of the actin in the resting cell is filamentous. The rest of the actin is soluble (60 percent) and is in a 1:1 complex with β4-thymosin. Filaments are stable because they are capped on their barbed ends by capZ. (Active) Shape change begins when calcium rises into the micromolar level and gelsolin becomes active. Gelsolin binds to actin filaments, interdigitates, and causes filaments to fragment. After fragmentation, gelsolin remains bound to the barbed filament end. Assembly of actin begins when capping proteins are dissociated from the barbed ends of the filament fragments formed in the rounding step by polyphosphoinositides (ppIs) and when the actin-related protein (ARP2/3) complex in platelets is activated to nucleate de novo filaments. Actin monomers, stored in complex with β4-thymosin, are the source of the actin for this polymerization event. Transfer of actin from β4-thymosin to the barbed ends of actin filaments is facilitated by profilin. Once assembly is complete, CapZ recaps the barbed filament ends. (Adapted with permission from Michelson A: Platelets. 2nd edition. Boston, MA: Academic Press/Elsevier; 2007.)
Filamin is a prototypical scaffolding protein that attracts binding partners, including the small guanosine triphosphatase (GTPase), RalA, Rac, Rho, and Cdc42,178 and positions them adjacent to the plasma membrane.179 Approximately 90 percent of the filamin in resting platelets interacts with the cytoplasmic tail of the GPIbα subunit of the GPIb-IX-V complex via a binding site in filamin’s second rod domain (repeats 17 to 20).180,181 This interaction has three consequences. First, it positions filamin’s self-association domain and associated partner proteins at the plasma membrane while presenting filamin’s actin-binding sites into the cytoplasm. Second, because a large fraction of filamin is also bound to actin, it aligns the GPIb-IX-V complexes into rows on the plasma membrane surface of the platelet over the underlying actin filaments. Third, because the filamin linkages between actin filaments and the GPIb-IX-V complex pass through the pores of the spectrin lattice, it restrains the molecular movement of the spectrin strands in this lattice and holds the lattice in compression. The filamin–GPIbα connection is essential for the formation and release of discoid platelets by megakaryocytes, as platelets lacking this connection are produced in lower numbers and the ones that are produced are abnormally large and fragile. Platelets deficient in GPIb (Bernard-Soulier syndrome; Chap. 10) are very large, perhaps as a result of abnormalities in organizing the cytoskeleton.
Platelets have sizable stores of glycogen that can often be seen on electron microscopy (see Fig. 2–2). Glycogen can be broken down into glucose 1-phosphate, and platelets can also take up glucose from their surrounding medium. Platelet glycolysis rates significantly exceed those of erythrocytes and skeletal muscle.182 Oxidative metabolism probably contributes to energy production in resting platelets, but it has been estimated that less than 1 percent of the pyruvate produced by glycolysis actually enters the citric acid cycle. The remainder is either converted to lactate or remains as pyruvate; both leave the platelet.183 Platelet mitochondria are capable of oxidation of fatty acids, but its importance to energy production is unclear.184–187 Platelets can actively metabolize acetate, which has been exploited to improve platelet storage conditions.185,188 Amino acids may also serve as energy sources and feed into the citric acid cycle, but their contributions are uncertain.
As in all cells, ATP consumption by platelets is partially devoted to maintaining ionic and osmotic homeostasis.189,190 In addition, the continuous polymerization and depolymerization of actin involves conversion of ATP to ADP, and this may account for as much as 40 percent of the ATP consumption in resting platelets.191 The continuous polymerization and depolymerization of tubulin that occurs in the coil of resting platelet involves conversion of guanosine triphosphate (GTP) to guanosine diphosphate (GDP), and thus consumes energy.159 Continuing dephosphorylation and rephosphorylation of phosphatidylinositols, which are important in signal transduction, has been estimated to consume as much as 7 percent of the total ATP produced.192 Protein phosphorylation also occurs as an ongoing process, but its fractional use of ATP is not clear in resting cells. Platelet stimulation leads to a marked increase in both glycolytic activity and oxidative ATP production, perhaps as a result of the abrupt decrease in ATP that occurs with platelet activation or the increase in cytoplasmic pH.187 The increased ATP appears to be used, at least in part, for phosphatidylinositide and protein phosphorylation.
Platelet stimulation is accompanied by a marked increase in both glycolytic activity and oxidative ATP production, perhaps through a feedback mechanism in response to the abrupt decrease in ATP that occurs with platelet activation or as a result of the increase in cytoplasmic pH.193 The increased ATP appears to be utilized, at least in part, in phosphoinositide phosphorylation and protein phosphorylation.
Peroxisomes In platelets, some of the main metabolic functions of peroxisomes include fatty acid β-oxidation, plasmalogen (a phospholipid) synthesis, and synthesis of platelet-activating factor (PAF).194 They contain acyl-CoA:dihydroxyacetone phosphate acyltransferase, which catalyzes the first step in the synthesis of ether-containing phospholipids. Deficiencies of this enzymatic activity have been identified in the cerebrorenal Zellweger syndrome, and the platelet activity can be used to diagnose the disorder.195,196
Mitochondria Platelets contain approximately four to seven mitochondria of relatively small size, often located near the plasma membrane; they are involved in oxidative energy metabolism.197–199 Control of mitochondrial Bcl-2 family proteins, including Bcl-x1 and Bak, directly affects a platelet’s life span, and alterations in these proteins can produce thrombocytopenia (Chaps. 1 and 7).200 Release of mitochondria upon platelet activation, either in microparticles or free in the circulation, may contribute to inflammation and nonhemolytic transfusion reactions.199 Abnormalities of mitochondrial enzymes, including the reduced form of nicotinamide adenine dinucleotide (NADH) coenzyme Q reductase (complex I), have been implicated in the pathophysiology of aging and several neurodegenerative disorders, including Alzheimer disease, schizophrenia, and some forms of Parkinson disease. Assays of platelet mitochondrial enzyme levels have been used in these studies.201–206 In addition, hyperglycemia-induced mitochondrial superoxide generation may contribute to the enhanced platelet aggregation observed in diabetes.207 Loss of the mitochondrial inner leaflet potential has been associated with surface expression of platelet procoagulant activity and coated platelet formation (see “Platelet Coagulant Activity” below).208–211
The cytoskeleton establishes the platelet’s native structure and its ability to respond to stimuli through changes in shape and force generation; as such, the cytoskeleton can be considered analogous to an animal’s bones and muscles. Table 2–1 lists the major components of the platelet contractile system. These elements are thought to contribute to platelet shape change, secretion, and clot retraction after platelet activation.
When exposed to a variety of agonists, platelets undergo dramatic changes in shape within seconds. Shape change follows a reproducible sequence of events during which the resting platelet cytoskeleton is dismantled and reorganized. The first noticeable change following activation is the dismantling of the microtubule coil and conversion from discs to spheres. Filopodia and lamellipodia, generated by new actin filament assembly, then extend from the plasma membrane. At the same time, intracellular organelles and granules and the dismantled microtubule coil are compressed into the center of the platelet. Once shape change is finished, the actin cytoskeleton is used as a platform for contraction, and contractile tension is exerted between platelets and between platelets and the adjacent fibrin strands.
Platelet shape change occurs in response to many different agonists. It involves loss of the platelet’s normal discoid shape (approximately 1.5 to 2.5 μm diameter and approximately 0.5 to 0.9 μm width) and transformation to a spiny sphere with long, thin filopodia extending several micrometers out from the platelet and ending in points that are as small as 0.1 μm in diameter (see Fig. 2–2).95,212 In the aggregometer, it has been generally assumed that the initial decrease in light transmission immediately after adding certain agonists is a reflection of platelets undergoing shape change,213 but this interpretation has been challenged by the suggestion that microaggregation, rather than shape change, accounts for this phenomenon.214 Although the reason platelets undergo shape change is unclear, one possibility is that it reduces electrostatic repulsion between two negatively charged platelets or between a platelet and a negatively charged surface or cell without the need to reduce surface charge density. Thus, after changing shape, the tip of a platelet filopodium can more easily approach and make contact with a surface or a cell because the great bulk of the repulsive surface charge is now at a distance from the tip.215
A change in platelet shape from disk to sphere is the first event that is observed as the platelet is activated. Agonist binding to select receptors activates phospholipase (PL) Cβ, which hydrolyzes membrane-bound PI-4,5-bisphosphate to inositol-1,4,5-triphosphate (IP3) and diacylglycerol. IP3 then binds to receptors on the DTS/sarcoplasmic reticulum, generating a rise in cytosolic calcium concentrations to 5 to 10 μM. While calcium can influence the activity of many actin-binding proteins, one of the major proteins that is activated is gelsolin, which is present in platelets at a concentration of approximately 5 μM. Actin filaments in resting platelets are relatively stable because their barbed ends (the end from which they can grow by adding additional actin monomers) are capped with the protein CapZ and α,γ-adducins (see Fig. 2–5). Calcium-activated gelsolin both severs existing actin filaments and caps the newly created barbed ends. This increases the number of actin filaments by an estimated 10-fold and substitutes gelsolin for CapZ and α,γ-adducins as the actin filament capping protein.216 Severing of actin filaments that interact with the planar lattice composed of filamin A (actin binding protein), GPIb/IX, and spectrin in the membrane cytoskeleton releases the constraints on the spectrin network. This allows the membrane skeleton to swell (but not produce filopodia) (see Fig. 2–5) by incorporating into the plasma membrane the membranes from the open canalicular system and, later, the membranes from the granules that release their contents.
The protrusive force for lamellipodia and filopodia formation comes from new actin polymerization, such that there is a doubling of actin filament content. This burst of actin filament assembly is powered by the generation of barbed-end nucleation sites after receptor activation. These nucleation sites are generated de novo by the activation of the Arp2/3 complex or by the exposure of the barbed ends of preexisting filaments.217 Because barbed ends have a higher affinity for actin molecules than do the actin sequestering proteins, they have the capacity to initiate actin filament polymerization.
Platelets contain two proteins whose main function is to bind and sequester actin monomers. The first is profilin, which is present at a concentration of 50 μM. Profilin can sequester actin monomers from the pointed ends of actin filaments, but not the barbed ends. Profilin also functions as a major transfer factor in actin filament polymerization. The second and more abundant protein involved in sequestration of actin monomers and stimulation of the polymerization of actin is thymosin-β4. With a platelet concentration of 55 mM, it is equimolar to actin. Thymosin-β4 binds actin molecules with an affinity that is greater than that of the pointed end of the actin filament, allowing it to compete effectively for molecules from the pointed end. Thymosin-β4 has a lower affinity for actin monomer than actin has for the barbed end of the filament, resulting in filament assembly when barbed ends are free. Thymosin-β4 maintains a large pool of unpolymerized actin, and 60 percent of the total actin in the platelet is bound to thymosin-β4. The affinity of thymosin-β4 for actin monomer is regulated by the nucleotide that is bound to actin.218
The platelet actin assembly reaction that follows the addition of agonists starts when free barbed ends are formed (see Fig. 2–5). Barbed ends are generated by the uncapping of filament ends and the de novo assembly of filaments by the Arp2/3 complex. Platelets contain high concentrations of barbed-end capping proteins that regulate the accessibility of these ends to regulate actin dynamics. Platelets contain 5 μM each of gelsolin219 and capZ,220 and 3 mM of adducin.221 Uncapping of the actin filaments appears to be accomplished by the inactivation of capping proteins by phosphoinositides that are produced during platelet activation, including PI-3,4-bisphosphate (PI3,4P2), PI4,5P2, and PI3,4,5P3.216 The uncapped actin filaments act as nuclei onto which actin monomers (which are maintained in an available pool by association with thymosin-β4) can assemble on the barbed ends of the filaments. Profilin accelerates actin polymerization by facilitating the transfer of actin from the actin-thymosin-β4 complex to the barbed ends of the actin filaments. In addition to exposing new filament ends as a source of nuclei, new nucleation sites are generated by activation by the Arp2/3 complex. The Arp2/3 complex mimics the pointed ends of actin filaments and stimulates barbed-end assembly of actin filaments. The Arp2/3 complex is made up of seven polypeptides, two of which have actin-related sequences, Arp2 and Arp3.222,223 Platelets contain high concentrations of the Arp2/3 complex (2 to 10 μM). Approximately 30 percent of the Arp2/3 complex is bound to the resting platelet cytoskeleton. Once platelets are activated, the Arp2/3 complex redistributes to the cytoskeleton, increasing three-fold and concentrating in the lamellipodial zone of actin filament assembly. Several signaling pathways regulate the activity of the Arp2/3 complex, including Wiskott-Aldrich syndrome protein (WASP) family members. Mutations in the WASP gene result in Wiskott-Aldrich syndrome, an inherited X-linked recessive disorder characterized by thrombocytopenia and T-cell immunodeficiency (see Chap. 11).
Simultaneous with these changes, the peripheral microtubule coil becomes constricted and fragmented and is ultimately compressed into the center of the cell. As the filopodia form, the platelet’s granules and organelles move to the center, surrounded by the microtubule coil, resulting in an increase in electron density. Activation of myosin II via phosphorylation of myosin light chain kinase contributes to the inward contractile force by its interaction with the actin fibers.
After platelets adhere to surfaces, they undergo variable degrees of spreading and activation. The patterns of spreading and activation depend primarily on the protein surface on which they spread, with collagen consistently inducing the most activation.224,225 In addition to the nature of the surface, the protein density, especially in the case of fibrinogen, can dramatically affect the signaling systems that are activated in the adherent platelets.226 Activation can result in release of granule contents and exposure of activated integrin αIIbβ3 receptors on the luminal surface of the platelets, where they are strategically located to bind adhesive glycoprotein ligands that can recruit additional platelets.227 If the surface density of platelets is sufficient, the platelets can also enter into lateral associations, which appear to depend on integrin αIIbβ3.228 In general, platelet spreading results in the development of broad lamellipodia rather than spike-like filopodia (see Fig. 2–2).216,229 The different morphologies of platelet spreading reflect differences in the organization of the network of actin filaments. Ultrastructural examination of lamellipodia reveals them to be replete with actin filaments that are organized into orthogonal networks. This organization is established by the actin filament crosslinking protein filamin A. In contrast, filopodia contain long actin filaments that are organized as tight bundles. These structural differences reflect the different signals initiated by the adhesion process, and both PIs and the small GTPase molecules Rac and Cdc42 appear to be particularly important in this process.154 In platelets, Rac is activated by thrombin receptor ligation, and it stimulates actin filament uncapping.230 Proteins that have been implicated in organizing the tips of the filopodia where the actin bundles attach to the plasma membrane are the small GTPase Cdc42, the exchange protein WASP, vinculin, vasodilator-stimulated protein (VASP), zyxin, and profilin.111 Pleckstrin, a platelet protein that is phosphorylated during platelet activation, appears to participate in this process by binding to PIs and affecting Rac via an exchange factor.231,232 Platelets from mice deficient in pleckstrin have a defect in granule secretion, integrin αIIbβ3 activation, and aggregation mediated by protein kinase C. Thrombin can overcome this abnormality via a pathway involving PI3K.233 Signaling after adhesion results from the assembly of protein complexes on the cytoplasmic surfaces of the receptor(s) involved in the adhesion process, including focal adhesion kinase (FAK), which is activated by integrin ligation and colocalizes with a number of cytoskeletal proteins. Deletion of FAK in megakaryocytes and platelets results in defects in platelet spreading.234 These complexes then initiate local cytoskeletal rearrangements as well as the generation of signaling molecules that act throughout the platelet to produce a variety of effects, including the translation of new proteins.235–238 The nature and extent of the signaling may determine whether the adherent platelets recruit additional platelets or white blood cells. In particular, the conversion of spread platelets to a microvesiculated procoagulant form has been associated with the recruitment of neutrophils.239 Additionally, spread platelets can assemble fibronectin matrix on their surface, which may be important in stabilizing platelet–platelet interactions.240
Membrane glycoproteins are affected by cytoskeletal rearrangements associated with platelet shape change and spreading. Activation of platelets in suspension under certain conditions results in movement of GPIb/IX receptors from the surface of platelets to the open canalicular system.241,242 With adherent platelets, the GPIb internalization is much slower.111 The initial effect of activation on integrin αIIbβ3 is an approximate doubling of these receptors on the plasma membranes, as preassembled receptors in α granules, and perhaps dense bodies and the open canalicular system, join the plasma membrane. Inside-out activation of integrin αIIbβ3 has been associated with cytoskeletal changes, in particular, the binding of talin to the integrin β3 cytoplasmic domain.243–246 Tyrosine kinases, including FAK33,247 and Src,247 may play a role in this process, along with cortactin, a protein of Mr 85 kDa that is phosphorylated on tyrosine, and small GTP binding proteins such as Rho, Rac, and Cdc42.216,229,248,249 When the attachment of integrin αIIbβ3 to the cytoskeleton includes actin and myosin, the force produced by the cytoskeleton on the integrin may supply the energy to produce the conformational changes that lead to higher ligand binding affinity.250 After activation, more integrin αIIbβ3 molecules become associated with the cytoskeleton, and this presumably reflects the interaction with talin and other cytoskeletal proteins and ligand-induced integrin clustering, resulting in the development of protein complexes, including cytoskeletal proteins, on the cytoplasmic surface of the receptor.237,245,251 When ligand-coated beads are added to adherent platelets and bind to integrin αIIbβ3 receptors, the beads are transported to the center of the platelets, indicating that the cytoskeleton can move integrin receptors that have bound ligand.252,253
Platelets contain calpains, which are calcium-dependent, sulfhydryl-containing, neutral proteases composed of two subunits that preferentially cleave cytoskeletal proteins, in particular filamins and talin,229,254 but have also been reported to cleave the cytoplasmic domain of integrin β3 and a number of molecules involved in signaling, including kinases and phosphatases (see “Calcium-Dependent Proteases [Calpains]” below). μ-Calpain requires micromolar calcium, and m-calpain requires millimolar calcium for activation. It has been proposed that calpains are involved in cytoskeletal reorganization upon platelet activation, specifically via cleavage of the integrin β3 cytoplasmic tail and talin upon ligand engagement.245,255–257 Calpain cleavage of the integrin β3 cytoplasmic tail may switch the function of the integrin from promoting platelet spreading to mediating clot retraction.258 Calpains have also been implicated in platelet spreading, microparticle formation, and the generation of platelet coagulant activity.229,256,259 Mice lacking μ-calpain have reduced platelet aggregation and clot retraction, but normal bleeding time.260
The contractile mechanism involving actin and myosin is thought to facilitate granule secretion, but the details remain obscure.261,262 In fact, mice with nearly complete disruption of the platelet heavy-chain myosin gene, Myh9, have a defect in secretion, but only in response to low concentrations of select agonists.263 The cytoskeleton of resting platelets consists of the membrane skeleton described above, which lies just beneath the membrane, and a lacy cytoplasmic actin filament network composed of 2000 to 5000 linear actin polymers that also contains α-actin, filamins (actin binding proteins) A and B, tropomyosin, vinculin, and caldesmon.176,177,248,249,264–268 The contractile response is also thought to be initiated by an increase in cytosolic calcium, which results in the formation of a calcium-calmodulin complex that then activates myosin light-chain kinase; phosphatases and cyclic adenosine monophosphate (cAMP) kinase can modulate this response. After the initial platelet shape change, actin becomes organized centrally into thick filamentous masses, where it probably associates with phosphorylated myosin filaments.269,270 The centralization of organelles within a contractile ring correlates with secretion.95 There is controversy, however, as to whether platelets secrete their granular contents by fusion with the open canalicular system in the center of the platelet or by direct fusion with the plasma membrane, or both.95,100
When blood initially clots in vitro, the fibrin mesh extends throughout, trapping virtually all of the serum in a gel-like state. If platelets are present, within minutes to hours, the clot retracts, extruding a very large fraction of the serum.271 This process is thought to mimic in vivo phenomena that result in consolidation of thrombi and perhaps enhancement of wound healing. Clot retraction has also been implicated in decreasing porosity and solute transport so as to concentrate intrathrombus thrombin,272 as well as decreasing the efficiency of thrombolysis, which may partially account for the resistance of platelet-rich thrombi to fibrinolytic agents.273 The platelet requirement for clot retraction is indisputable, as is a requirement for integrin αIIbβ3 and a contractile mechanism involving actin and myosin.274,275 In fact, nearly complete selective disruption of the myosin Myh9 gene in murine megakaryocytes gives rise to a phenotype characterized by macrothrombocytopenia; absence of clot retraction; reduced secretion in response to low concentrations of agonists, but not high concentrations; prolonged bleeding time; and protection from thrombus formation.263 The mice do not, however, spontaneously bleed.263 Myosin activation involves phosphorylation of the myosin light chain, a process that is governed by calcium-regulated myosin light-chain kinase activity and Rho kinase–regulated myosin phosphatase activity. Calpain cleavage of the cytoplasmic tail of integrin β3 may promote RhoA activity and serve as a molecular switch to convert platelet spreading to clot retraction.258 Other signaling molecules appear to contribute to clot retraction, including the Eph kinase EphB2,276 protein phosphatase 2B,277 and PI3K.278 Despite these data, no model describing the details of the clot retraction process has gained acceptance.279 Proposed mechanisms include movement of platelet filopodia along fibrin strands, tugging of fibrin strands by filopodia, and internalization of fibrin by the action of the membrane skeleton.274,275,279–282
Platelet integrin αIIbβ3 is required for clot retraction, as demonstrated by studies of patients with Glanzmann thrombasthenia (Chap. 11) and studies of normal platelets in the presence of agents that block either the integrin αIIbβ3 receptor280,283–288 or the fibrinogen γ-chain C-terminal sequence that mediates interactions with the integrin.289 It also requires disulfide bond exchange290 and the tyrosine residues on the integrin β3 subunit that are phosphorylated upon platelet activation and contribute to outside-in signaling.291 Clot retraction correlates temporally with an integrin αIIbβ3-dependent decrease in protein tyrosine phosphorylation, presumably via activation of one or more phosphatases,292 and may require both integrin-mediated mitogen-activated protein kinase (MAPK) activation293 and translation of proteins such as Bcl-3, with the latter facilitated by ligand binding to integrin αIIbβ3.294 However, results with integrin αIIbβ3 antagonists demonstrate differences in their ability to inhibit clot retraction that do not correlate with their ability to block fibrinogen binding to platelets,280,287 and patients with Glanzmann thrombasthenia differ in the extent of their defect in clot retraction. Some integrin αIIbβ3 mutations, such as integrin β3 L262P, interfere with interactions with fibrinogen but do not prevent interactions with fibrin and clot retraction.295 Of particular note, fibrinogen lacking the γ-chain C-terminal sequence (amino acids 400 to 411) that mediates binding to platelet integrin αIIbβ3, as well as the two Arg-Gly-Asp (RGD)-containing regions in fibrinogen, is still capable of supporting clot retraction.296,297 It is well established that when fibrinogen converts to fibrin, new sites become exposed on the surface of the molecule. Therefore, one possible explanation for this paradox is that additional or alternative integrin binding sequences in the fibrinogen γ-chain (e.g., 316 to 322, 370 to 383, or other regions) may be able to mediate clot retraction.298,299 Potential binding sites for the γ370 to 381 sequence, which is better expressed on fibrin than fibrinogen, on the integrin αIIb β-propeller region, were identified, and peptides from these regions inhibit clot retraction.300 Factor XIII also plays an important role in clot retraction; it has been proposed to mediate the translocation of the fibrinogen/fibrin–integrin αIIbβ3 complex to sphingomyelin-rich lipid rafts in the platelet membrane as well as crosslink the complex to cytoskeletal and contractile elements.301,302 It is also possible that GPIb/IX contributes to clot retraction by virtue of the binding of GPIbα to the thrombin and/or VWF bound to the fibrin.303,304 Thus, although integrin αIIbβ3 is required for clot retraction, the process is not a simple reflection of fibrinogen binding to integrin αIIbβ3.
Platelets possess secretory granules and mechanisms for cargo release to amplify responses to stimuli and influence the surrounding environment. Platelet granule structures include α and dense granules, lysosomes, and peroxisomes.
Lysosomes are produced from the endosomal membrane system through a complex mechanism involving membrane and protein sorting and trafficking.305 Platelet lysosomes contain acid hydrolases typical of these organelles (e.g., β-glucuronidase, cathepsins, aryl sulfatase, β-hexosaminidase, β-galactosidase, endoglucosidase [heparitinase], β-glycerophosphatase, elastase, and collagenase).197 With activation, platelets secrete some of these enzymes; however, lysosomal contents are more slowly and less completely released than are those from α granules and dense bodies.306–308 Thus, stronger agonists are required to induce lysosomal enzyme release than release from the other granules, and their appearance on the platelet plasma membrane serves as a marker of high-level platelet activation.309,310 The elastase and collagenase activities released from platelet lysosomes may contribute to vascular damage at sites of platelet thrombus formation.311 The heparitinase may be able to cleave heparin-like molecules from the surface of endothelial cells, and the resulting soluble molecules appear to inhibit growth of smooth muscle cells.312
Platelets contain approximately three to eight electron-dense organelles, 20 to 30 nm in diameter (see Fig. 2–2).76,262 The intrinsic electron density of dense bodies when viewed as unstained whole mounts derives from their high content of calcium76,197; the granules are also dense when viewed by transmission electron microscopy because they are highly osmophilic.262 Dense granules contain high concentrations of serotonin, which is taken up from plasma by a plasma membrane carrier and then trapped in the dense bodies.262 Trapping of serotonin may occur as a result of the lower pH (approximately 6.1) maintained in dense granules as a result of the action of a proton pumping ATPase on the dense-body membrane.262 ADP and ATP are also highly concentrated in dense bodies.197 There is more ADP than ATP in the dense bodies (ATP to ADP ratio = 2:3), which is the reverse of their relative concentrations in the cytoplasm (ATP to ADP ratio = 8:1). As there is little connection between the pools of adenine nucleotides in the cytoplasm and the dense bodies, they have been respectively designated as the metabolic and storage pools of adenine nucleotides.197 Storage of adenine nucleotides at such a high concentration in dense bodies appears to be achieved by stacking the ATP and ADP purine rings vertically in aggregates that are stabilized by the interactions of calcium ions with the polyphosphate groups.313,314 The planar hydroxyindole rings of serotonin may also enter these stacks, providing a molecular basis for the trapping mechanism. Trapping of serotonin must differ from that of adenine nucleotides, however, because dense granule serotonin exchanges readily with external serotonin.197 Transport and delivery of platelet-derived serotonin may play an important role in a variety of biologic phenomena including vasospasm, platelet coagulant activity, and liver regeneration.315
The membrane of dense granules contains glycoproteins that are also found on the plasma membrane and the membranes of α granules and lysosomes, including CD36, LAMP-2, CD63, P-selectin, αIIbβ3, and GPIb/IX. Abnormalities of eight different genes have been implicated in the Hermansky-Pudlak syndrome (HPS) (Chap. 11), an autosomal disorder characterized by a deficiency of dense bodies, and so these genes are presumed to participate in dense body formation. As with lysosomes, dense bodies are thought to derive from endosomes, via different types of multivesicular bodies (MVBs). The eight genes associated with HPS are thought to affect sorting and/or trafficking of membrane structures through participation in protein complexes that mediate these phenomena.316,317 These complexes include three biogenesis of lysosome-related organelles complexes (BLOCs) and the activator protein 3 (AP3) complex.305 Similarly, the product of the LYST gene, which is abnormal in some patients with Chédiak-Higashi syndrome (who also have abnormal dense bodies), has been proposed to associate with the dense granule membrane (Chap. 11).318 The LYST gene product may associate with the AP3 complex.305
The abnormalities of in vitro platelet function in patients with HPS suggest that released dense granule contents contribute to platelet activation through a positive feedback mechanism. Release of ADP, which is a potent platelet activator, and serotonin, a weaker agonist (see section “Signaling Pathways in Platelets” below), probably accounts for most of the positive feedback effects on platelet aggregation. ATP is a partial antagonist of ADP-induced activation, but as ATP is rapidly catabolized to ADP in plasma (T1/2 = 1.5 min), and ADP is rapidly catabolized to AMP (T1/2 = 4 min) and then to adenosine,197 a platelet inhibitor,319 it is difficult to predict the overall effect of ATP release. Adding to the complexity in vivo is the presence of an ecto-ADPase (CD39; ecto-ADPase) present on endothelial and lymphoid cells, which can metabolize ATP and ADP to AMP and thus probably limits the amount of ADP present.74 ATP released from platelets may also serve as a high-energy phosphate source for platelet ecto-protein kinases, which can phosphorylate several proteins, including CD36 (GPIV).320–322
An important platelet function is storage and release of a variety of bioactive substances packaged in α granules. α Granules are the most abundant granule type of platelets, numbering approximately 50 to 80 per platelet.323,324 They are approximately 200 nm in diameter on cross-section and demonstrate internal variation in electron density, often with an eccentric area of accentuated electron density, termed a nucleoid, in which β-thromboglobulin, platelet factor 4 (PF4), and proteoglycans are concentrated (see Fig. 2–2).325 The more electron-lucent areas contain tubular elements in which VWF, multimerin, and factor V are preferentially localized.76 Proteomic analysis of the releasate of activated human platelets has identified more than 300 proteins, most of which are stored within α granules.326–328 The list of α-granule proteins includes adhesive proteins, coagulation factors, protease inhibitors, chemokines, and angiogenesis regulatory proteins. Some of the most important proteins present in α granules are described in detail below. Platelets contain distinct subpopulations of α granules that undergo differential release of α-granule cargo during activation. For example, some α granules contain proangiogenic proteins, such as vascular endothelial growth factor (VEGF), whereas others contain antiangiogenic factors, such as endostatin (Fig. 2–6).329 These two subclasses of α granules can be differentially induced to undergo degranulation by exposure of human platelets to agonists specific for either protease-activated receptor (PAR)-1 or PAR-4. Fibrinogen and VWF are localized in separate α granules,330 and glass activation of platelets results in the selective release of the fibrinogen-containing granules.
Figure 2–6.
Platelets contain separate and distinct α-granule populations. A, B, and C. Specific pro- and antiangiogenic regulators organize into separate, distinct α granules in resting platelets. Double immunofluorescence microscopy of resting platelets using antibodies against vascular endothelial growth factor (VEGF) (A) and endostatin (B) and an overlay (C). D. Localization of proteins in resting, human platelets using immunoelectron microscopy of ultrathin cryosections. Double immunogold labeling on platelet sections was performed with the use of anti-VEGF antibody and antiendostatin antibodies. Large gold particles representing anti-VEGF staining (15 nm, arrows) are evident on one population of α granules, and small gold particles (5 nm) representing endostatin staining are abundantly present on a different population of α granules (arrowheads). E, F, and G. Pro- and antiangiogenic regulatory proteins are also segregated into separate, distinct α granules in megakaryocyte proplatelets. Megakaryocytes generate platelets by remodeling their cytoplasm into long proplatelet extensions, which serve as assembly lines for platelet production. Distinct α granules are visualized along proplatelets. Shown is a double immunofluorescence microscopy experiment of proplatelets using antibodies against VEGF (E) and endostatin (F), and an overlay (G). (Reproduced with permission from Italiano JE, et al: Angiogenesis is regulated by a novel mechanism: pro- and antiangiogenic proteins are organized into separate platelet alpha granules and differentially released, Blood. 2008 Feb 1;111(3):1227–1233.)
The α granule acquires its protein content by both biosynthesis (predominantly at the megakaryocyte level) and endocytosis (at both the megakaryocyte and circulating platelet levels). Small amounts of virtually all plasma proteins are nonspecifically taken up into α granules, and thus, the plasma levels of these proteins determine their platelet levels.331,332 For example, the α-granule pool of immunoglobulins contains most of the platelet immunoglobulin; therefore, total platelet immunoglobulin is more affected by changes in plasma immunoglobulin levels than by changes in surface immunoglobulin.331,332
The cell biologic pathways that regulate α-granule assembly are not fully understood, but several studies suggest MVBs play a crucial intermediary role in α-granule biogenesis.316,333 These membranous sacs, containing numerous small vesicles, develop from budding vesicles in the Golgi complex within megakaryocytes and can interact with endocytic vesicles. They are abundant in immature megakaryocytes and decrease in number with cellular maturation, suggesting that they are the precursors of α granules and/or dense bodies. MVBs may also function as a sorting hub to rout proteins into distinct classes of α granules.
The platelet-specific proteins (PF4 and the β-thromboglobulin family) are present in α granules at concentrations that are approximately 20,000 times higher than their plasma concentrations (when each is expressed as a fraction of total protein in platelets or plasma, respectively).334,335 These Mr 7000 to 11,000 proteins all bind to heparin, but with varying affinities. They also share amino acid sequence homology with each other and with other members of the “intercrine-cytokine” family of molecules, such as interleukin (IL)-8 (neutrophil-activating peptide 1 [NAP1]), which are active in inflammation, cell growth, and malignant transformation.336–338
PF4 is a CXC chemokine (CXCL4) that does not contain the Glu-Leu-Arg (ELR) conserved sequence.339,340 It binds to heparin with high affinity and can neutralize heparin’s anticoagulant activity.335,341–343 PF4 tetramers complex with a proteoglycan carrier.344,345 Specific PF4 lysine residues (amino acids 61, 62, 65, and 66) are implicated in its binding to heparin, and x-ray crystallography indicates that these lysines are on the surface of the PF4 tetramer and interact with negatively charged heparin molecules that wind around this core.346–348
After PF4 is released from platelets, it binds to heparin-like molecules on the surface of endothelial cells.346 Heparin administration can mobilize this endothelial-bound pool of PF4 into the circulation.346 PF4-heparin complexes and PF4-heparin-like molecule complexes on endothelial cells have been implicated as the target antigens in heparin-induced thrombocytopenia with thrombosis.349,350 PF4 also binds to hepatocytes, which take it up and catabolize it.351 PF4 is a weak neutrophil and fibroblast attractant.340,352 It inhibits angiogenesis, perhaps through inhibition of endothelial cell proliferation.353 A large number of other activities have been ascribed to PF4, including histamine release from basophils354; inhibition of both tumor growth353 and megakaryocyte maturation355–357; reversal of immunosuppression352,358; enhancement of fibroblast attachment to substrata359; potentiation of platelet aggregation360; inhibition of contact activation361; and enhancement of both polymorphonuclear leukocyte responsiveness to the activating peptide f-Met-Leu-Phe and monocyte responsiveness to lipopolysaccharide.362,363
The β-thromboglobulin family of proteins are CXC chemokines that contain the conserved Glu-Leu-Arg (ELR) sequence.340 They include platelet basic protein, low-affinity PF4 (connective tissue-activating peptide III [CTAP-III]), β-thromboglobulin, and β-thromboglobulin-F (NAP2, CXCL7).334,364–366 All of these proteins share the same carboxy terminus but differ in the length of their amino termini, presumably as a result of proteolytic digestion of the parent molecule, platelet basic protein. These proteins bind to heparin but with lower affinity than PF4, and thus neutralize heparin less well. Unlike PF4, they are cleared from the circulation by the kidney rather than the liver.367 CTAP-III is a weak fibroblast mitogen, and β-thromboglobulin is a chemoattractant for fibroblasts.340 β-Thromboglobulin-F NAP2 (CXCL7) binds to CXCR2 and is chemotactic for granulocytes and activates them to undergo endocytosis.339,340,366 Platelet α granules also contain additional chemokines that can variably activate leukocytes and platelets.339
The biochemistry of the adhesive glycoproteins contained in α granules and others variably present in plasma and extracellular matrix is described in Table 2–2 and in other chapters (e.g., Chaps. 3 and 15 for fibrinogen and Chap. 16 for VWF). Their relative concentrations in α granules varies significantly. Their localization in platelet α granules allows them to achieve high local concentrations when released from platelets at the site of vascular injury.
| Protein | Subunit, kDa | Unusual 1° Structural Features & Modifications | Domain Homologies & Binding Regions | Mature Protein Composition | Mature Protein Mr | Known Interactions | Known Platelet Receptors | Electron Microscopy Structure | Plasma Concentration, mcg/mL | Platelet Concentration,* mcg/mL | Ratio Platelet/Plasma | Sites of Synthesis |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Collagens | 95–180 | Gly-Pro-X repeating sequence Hydroxylysine Hydroxyproline | RGD Right-handed triple helix | Tropocollagen = 3 chains | Variable Thrombospondin | α2β1 (GPIa/IIa; CD49b/CD29; VLA-2) GPVI GPIV (CD36)? | Tropocollagen = rodlike coil, 15 × 3000; other forms have variable degrees of fibril formation | – | – | – | Fibroblasts | |
| Type I | α1(I) α2(I) | DGEA† VWFC | [α1(I)]2α2(I) (major component) [α1(I)]3 | Fibronectin von Willebrand factor | ||||||||
| Type III | α1(III) | VWFC | [α1(III)]3 | |||||||||
| Type VI | α1(VI) α2(VI) α3(VI) | 3 VWFA 3 VWFA 12 VWFA | α1(VI)α2(VI)α3(VI) | |||||||||
| von Willebrand factor | 220 (2050 amino acids) | Large propeptide (741 amino acids); A, B, C, D, E repeats | αIIbβ3 – RGD 1789–1791 I Domains GPlb – 230–310 | Dimer = protomer Multimers of protomers from 2–~40 via disulfide bonds | 880,000– ~20,000,000 | Collagen Heparin Factor VIII Fibrin | GPIb (CD42b, c) αIIbβ3 (GPIIb/IIIa; CD41/CD61) | Elliptical, nodular coil, length 5000, but with some 11,000 Å | 10 | 34 | 3.4 | Endothelial cells Megakaryocytes |
| Fibrinogen | Aα = 63 (625 amino acids) Bβ = 56 (461 amino acids) γ = 47 (427 amino acids) | Alternately spliced γ chains Phosphorylation of Aα | 2 RGDs in Aα (95–97 and 572–574) αVβ3 – RGD 572–574 αIIbβ3–C-terminal γ – chain dodecamer (400–411) | 2 Aα, 2 Bβ, 2 γ via disulfide bonds | 340,000 | Thrombospondin ?Collagen Staphylococci Factor XIII Thrombin | αIIbβ3 (GPIIb/IIIa; CD41/CD61) αVβ3 (CD51/CD61) | Trinodular, asymmetrical; 475 Å diameter | 3000 | 7300 | 2.4 | Hepatocytes |
| Vitronectin | 1 chain = 75 (458 amino acids) 2 chain = 65 + 10 via disulfide bonds | Met → Thr polymorphism | RGD Somatomedin B 2 Hemopexin | Same as subunits | 75,000 and 65,000+10,000 | Glass Plastic Heparin Serine protease: serpin complexes PAI-1 uPAR Factor XIII | αIIbβ3 (GPIIb/IIIa; CD41/CD61) αVβ3 (CD51/CD61) | 350 | 800 | 2.3 | ?Hepatocytes | |
| Fibronectin | 220 (2355 amino acids) | Types I, II, and III repeats Alternately spliced forms | RGD (1493–1495) | Heterodimer via disulfide bonds | 440,000 | Fibrin Heparin Collagen DNA Staphylococci | α5β1 (GPIc*/IIa (CD49e/CD29; VLA-5) αIIbβ3 (GPIIb/IIIa; CD41/CD61) | Extended antiparallel dimeric structure | 300 | 315 | 1.1 | Hepatocytes Fibroblasts ?Endothelial cells Megakaryocytes Monocytes, etc. |
| Thrombospondin 1 | 180 (1150 amino acids) | RGD (?functional) VTCG† α1(I) Collagen Epidermal growth factor Malaria antigen | Trimer via disulfide bonds | 450,000 | Calcium Plasminogen Collagen Fibrinogen Histidine-rich glycoprotein Fibronectin Laminin Heparin | GPIV (CD36) αIIbβ3 (GPIIb/IIIa; CD41/CD61)? Integrin associated protein (CD47) | 3 Asymmetrical dumbbells, joined near smaller globular domains | 0.16 | 4900 | 30,625 | Megakaryocytes Many cultured cells | |
| Osteopontin | 32 (298 amino acids) | Phosphorylation Sulfation | RGD | Hydroxyapatite Plaque components | αVβ3 | – | – | – | Bone ?Other cells | |||
| Laminin | A = 400 B1 = 215 (1765 amino acids) B2 = 205 (1576 amino acids) | YIGSR† RGD (?functional) EGF | A, B1, B2, via disulfide bonds | 850,000 | Collagen type IV Nidogen/entactin Osteonectin Heparin sulfate C1q Plasminogen Plasmin | α6β1 (GPIc/IIa; CD49/CD29; VLA-6) | Cross-like structure | – | – | – | Fibroblasts Many other cell types | |
| Multimerin | 155 or 167 kDa | Large prepro-peptide (1228 amino acids) | RGD in N-terminal region EGF | 450,000– ~5,000,000 | Factor V | Unknown | Unknown | – | – | – | Megakaryocytes Endothelial cells |
Multimerin comprises a family of disulfide-linked homomultimers, ranging in molecular weight from 450,000 to many millions.368 The Mr 450,000 multimer is thought to be a trimer of a single subunit of either Mr 167,000369 or Mr 155,000368 that is synthesized in megakaryocytes and endothelial cells and stored in the electron-lucent region of α granules in platelets and dense-core granules in endothelial cells.370 It colocalizes with VWF in platelets, but not endothelial cells. Although multimerin’s multimeric structure is similar to that of VWF, the deduced amino acid sequence of its subunit is not homologous to that of VWF.368 The prepromultimerin subunit contains 1228 amino acids. It undergoes glycosylation and proteolysis during synthesis. It is composed of a number of domains, including an aminoterminal region that includes an RGD sequence, coiled coil sequences, epidermal growth factor (EGF)-like domains, and a carboxyterminal globular head similar to that found in the complement protein C1q. Multimerin binds both factor V and factor Va, and all of the biologically active factor V in platelets is bound to multimerin.325 With thrombin activation of platelets, factor V separates from multimerin, and the higher molecular weight multimerin multimers bind to platelets. Multimerin does not circulate in plasma at an appreciable concentration, but it may act as an adhesive extracellular matrix protein.
Fibrinogen is concentrated in α granules as judged by the ratio of platelet-to-plasma fibrinogen. Megakaryocytes do not appear to synthesize fibrinogen; instead, it is taken up from plasma by a process that involves the αIIbβ3 receptor.371 Because fibrinogen molecules that contain altered sequences in the γ chain are not stored in α granules, even when the molecules are heterodimeric (i.e., contain one normal and one abnormal γ chain), it is possible that uptake requires simultaneous binding of a single fibrinogen molecule to two different αIIbβ3 receptors via the γ-chain carboxyterminal sequence (see αIIbβ3 in the section “Platelet Membrane Glycoproteins” below and Chap. 11).371,372
The VWF stored in platelet α granules appears to contribute to hemostasis because in certain pathologic states it correlates better with bleeding symptoms than does plasma VWF concentration (Chap. 16). VWF is synthesized in megakaryocytes and endothelial cells. The multimeric structure of platelet VWF is thought to reflect endothelial VWF more nearly than plasma VWF, as higher Mr multimers are present.
Fibronectin is present in α granules, but no clear role in platelet function under normal conditions has been identified for this adhesive protein. Paradoxically, in murine models, fibronectin has been reported to both support platelet thrombus formation and inhibit platelet aggregation and thrombus formation41,373; the former effect may be mediated by insoluble fibronectin fibrils, whereas the latter may be mediated by soluble fibronectin.374
Vitronectin, which gets its name from its propensity to bind to glass, also binds to PAI-1, the urokinase receptor (uPAR), collagen, and heparin; it also forms ternary complexes with serine proteases and serpins in the coagulation and complement systems. It is present in platelets at levels that suggest it is concentrated,375 but it does not appear to be synthesized in megakaryocytes. The binding of PAI-1 with vitronectin stabilizes PAI-1 in its active conformation, and it has been proposed that only the approximately 5 percent of PAI-1 complexed with vitronectin in platelet α granules is active.150 Mice deficient in vitronectin have been reported to be protected from, or have a predisposition to develop, thrombosis, depending on the method of inducing thrombosis.376–378
Thrombospondin-1 is unique among the adhesive glycoproteins in blood in that it is present almost exclusively inside the platelet.379–381 It constitutes approximately 20 percent of the released platelet proteins. Thrombospondin-1 is synthesized by megakaryocytes, cultured endothelial cells, and other cultured cells.382,383 Although integrin αIIbβ3, GPIb/IX, integrin αVβ3, proteoglycans, integrin-associated protein (CD47 or IAP), and CD36 (GPIV) have all been implicated as receptors for thrombospondin,384–390 CD47 appears to be most important in initiating platelet activation by thrombospondin (see “Signaling Pathways in Platelet Activation and Aggregation” below).386,387,391 The phosphorylation state of CD36 (GPIV) may affect its ability to bind thrombospondin.385 Thrombospondin contains an Arginine-Glycine-Aspartate (RGD) sequence, which may contribute to its binding to platelets, but other regions are probably also involved.381,392 The conformation of thrombospondin varies with the calcium concentration of the surrounding environment. Thrombospondin can interact with many other adhesive glycoproteins, including fibronectin and fibrinogen,210,393,394 and it is a component of the extracellular matrix.395 Thrombospondin appears to stabilize platelet aggregates that are formed396; it may also act as a negative regulator of angiogenesis, modulate fibrinolysis, and contribute to activation of latent transforming growth factor (TGF)-β1 released from platelets (see below in this section).397,398
Platelets contribute approximately 20 percent of the factor V present in whole blood, with nearly all of it in α granules.399–401 Human platelet factor V appears to be taken up from plasma rather than being synthesized in megakaryocytes, which is in stark contrast to the situation in mice. When stored in α granules, factor V associates with multimerin.402,403 Platelet-derived factor V appears to undergo unique posttranslational modifications and proteolytic activation, resulting in resistance to protein C-catalyzed inactivation.404–406 Evidence from patients with inhibitors and deficiencies of plasma and platelet factor V indicate that platelet-derived factor V has an important role in hemostasis.399,407,408 Platelets undergo microvesiculation when activated, and the microvesicles, which are rich in factor V, are potent promoters of coagulation.409
Protein S (Chap. 4), plasminogen activator-1 (Chap. 25), and α2-plasmin inhibitor (Chap. 25) are also contained in α granules and can be released from platelets. Similarly, tissue factor pathway inhibitor (TFPI; Chap. 4), α1-protease inhibitor, and C-1 inhibitor (Chap. 4) have also been identified in α granules.
Gas6 is a 75-kDa vitamin K-dependent protein that contains γ-carboxyglutamic acids and is similar in structure to protein S.410,411 Gas6 was originally isolated as a growth arrest-specific gene from quiescent fibroblasts but subsequently was found to enhance platelet aggregation and secretion in response to several agonists.412 Mice deficient in Gas6 have abnormalities in platelet aggregation and are protected from experimental thrombosis.412 Gas6 is present in α granules and secreted with platelet activation. Platelets also express Mer, a tyrosine kinase receptor for Gas6, and mice deficient in Mer demonstrate both abnormalities in platelet aggregation and protection from thrombosis, but not to the same extent as mice deficient in Gas6.413,414 Other Gas6 receptors in the same family as Mer also appear to contribute to platelet thrombus stability.413–417
Platelet-derived growth factor (PDGF) is a disulfide-linked dimeric molecule of approximately Mr 30,000 that is mitogenic for smooth muscle cells.418 Platelet α granules contain a mixture of the homodimer PDGF-BB (30 percent) and the heterodimer PDGF-AB (70 percent); the different forms appear to have different functional activities.419 PDGF may play a role in normal cell proliferation, as well as in the development of atherosclerosis, tumor growth, wound repair, and fibroproliferative responses.420–422 After it was discovered in platelets and termed PDGF, other tissues were found to produce the same factor; thus, despite its name, PDGF is not exclusively derived from platelets. PDGF is structurally related to the transforming protein p28sis of simian sarcoma virus,423,424 and its receptor is in the tyrosine kinase family.425 Recombinant human PDGF-BB (becaplermin) is approved as adjunctive therapy to improve healing of foot ulcerations in diabetics.426
Platelets contain high concentrations of VEGF, an important stimulator of angiogenesis, and can release VEGF after stimulation in vitro and during the hemostatic response to a bleeding time wound.427–429 Megakaryocytes express mRNA of the three VEGF isoforms (121, 165, and 189 amino acids),430 and by immunoblot, VEGF protein bands of apparent molecular weights 34,000 and 44,000 are identifiable in platelets.431 Platelets and megakaryocytes also express the gene transcript for the VEGF receptor termed KDR.432 Another endothelial growth factor structurally related to VEGF, VEGF-C, has also been identified in platelets.433 Platelet levels of VEGF have been reported to be increased in malignancies, and so elevated levels of platelet VEGF may be a cancer biomarker.434,435 Platelet VEGF has also been postulated to play a role in tumor growth436 and proliferative retinopathy in sickle cell disease.437,438
EGF has also been identified in platelets, but the kinetics of its release upon thrombin or collagen stimulation differs from that of other granule proteins.439
Platelets contain the highest levels of all peripheral tissues of amyloid precursor protein (APP), which contains the sequence for the self-aggregating 40- to 43-amino-acid-residue peptide, Aβ, that has been strongly implicated in the pathogenesis of Alzheimer disease.440,441 The isoforms containing the Kunitz protease inhibitor domain (APP 770 and APP 751) predominate in platelets. Although synthesized as a membrane protein, platelet APP is cleaved by α-, β-, and γ-secretase activities, producing all of the fragments produced by neurons, as well as the soluble sAPPα, sAPPβ, and Aβ peptides, and the corresponding remaining C-terminal membrane-associated fragments.440,442,443 Calpain, which is present in platelets, can also cleave platelet APP.444 Approximately 90 percent of platelet APP is soluble and stored in α granules, but full-length APP surface expression is increased threefold by thrombin stimulation.445 Platelets are the major source of plasma sAPPs and Aβ.443,446 APPs released by platelets are potent inhibitors of factor XIa447 and IXa,448,449 and also can inhibit platelet aggregation induced by ADP or epinephrine. In contrast, Aβ appears to enhance ADP-induced platelet aggregation and support platelet adhesion. It is possible, but not certain, that plasma Aβ contributes to brain Aβ in Alzheimer disease.441 Patients with Alzheimer disease have been reported to display altered platelet APP metabolism.450–455
Factor XIII is present in the cytoplasm of platelets; it differs from plasma factor XIII in having only the “a” subunits (Chap. 3).456–459 Platelet factor XIII accounts for approximately 50 percent of total blood factor XIII,456,457 and platelet factor XIII may contribute to the plasma pool.460 Upon platelet activation, factor XIII redistributes to the platelet periphery where it associates with the cytoskeleton and crosslinks filamin and vinculin.461 It may also crosslink thymosin β4 to fibrin after thrombin stimulation462 and, in concert with calpain, decrease integrin αIIbβ3 adhesive function in thrombus formation on collagen.463 Transglutaminase-mediated conjugation of serotonin to α-granule proteins after platelet stimulation with collagen and thrombin results in the generation of a subpopulation of platelets that are coated with fibrinogen, thrombospondin, factor V, VWF, and fibronectin, either directly through ligand–receptor interactions or through interactions between the serotonin conjugates and platelet surface fibrinogen or thrombospondin (COAT platelets).464,465
Platelet α granules contain a high concentration of TGF-β1, an Mr 25,000 homodimeric protein that promotes the growth of certain cells and inhibits the growth of others.466–469 For example, TGF-β can increase thrombopoietin production by marrow stromal cells. In turn, thrombopoietin induces both increased megakaryocyte production and megakaryocyte expression of TGF-β receptors. The interaction of TGF-β with these receptors then results in inhibition of megakaryocyte maturation.470 TGF-β1 also induces synthesis of extracellular matrix proteins, PAI-1, and metalloproteinases. It has been implicated in wound healing, malignancy, and tissue fibrosis.471 In addition, TGF-β1 has been reported to enhance platelet aggregation through a nontranscriptional effect.472 Migration of endothelial cells is inhibited by TGF-β1, but it acts as a chemoattractant for monocytes and fibroblasts. TGF-β exists in three isoforms (TGF-β1, TGF-β2, and TGF-β3), but platelets contain only TGF-β1. TGF-β1 released from platelets can stimulate smooth muscle cells to express and release VEGF, thus perhaps supporting reendothelialization after vascular injury.473
TGF-β1 released from platelets is inactive (latent) because it is complexed with the remaining portion of its precursor protein (latency-associated peptide [LAP]).474 LAP, in turn, is covalently coupled to another protein, the latent TGF-β–binding protein-1 (LTBP-1), which localizes the complex to the extracellular matrix.475 Activation of latent TGF-β1 is a complex process that is thought to involve a conformational change in LAP that results in altering its ability to shield the active site in TGF-β1.475 Activation of latent TGF-β1 can be achieved by several different mechanisms, including acidification; proteolysis by plasmin, a furin-like enzyme, or other enzymes; traction produced by LTBP-1 binding to extracellular matrix and LAP interaction with integrin αVβ6 or αVβ8; interaction with thrombospondin-1 or a small peptide derived from thrombospondin-1; or exposure to stirring or shear.475–479 The interaction of LAP with integrin receptors via its RGD sequence probably plays a dominant role as mice with a mutation in this sequence have a phenotype like that of TGF-β1 null mice.480 The ability of thrombospondin-1 to activate TGF-β1 is of special interest because both TGF-β1 and thrombospondin-1 are present in α granules. However, data from mice suggest a minor role for platelet thrombospondin in either TGF-β1 packaging or activation.481–483 Only a very small percentage of the TGF-β1 released from platelets with thrombin stimulation becomes activated, but this amount is sufficient to activate synthesis of PAI-1.479,481,482,484 Based on animals models, TGF-β1 released from platelets has been implicated in promoting tumor metastases and cardiac fibrosis in response to constriction of the aorta or aortic valve stenosis.485–487 Active TGF-β can bind to three different cell surface proteins, a proteoglycan (β-glycan), and two serine/threonine kinases.471,485–488
Platelets may also release proteins that affect the uptake of oxidized low-density lipoproteins by macrophages, furnishing another potential link between platelet activation and atherosclerosis.489
In addition to the contents of α granules, activated platelets release both microparticles (see “Platelet Coagulant Activity” below), which are derived from the plasma membrane, and exosomes, which are internal membrane MVBs.490 Exosomes are smaller than microparticles (40 to 100 nm vs. 100 to 1000 nm), enriched in CD63 and tetraspanins (see section “Platelet Membrane Glycoproteins” below), and relatively deficient in membrane proteins such as GPIb/IX and platelet-endothelial cell adhesion molecule (PECAM)-1. Unlike microparticles, exosomes are not highly procoagulant as judged by their inability to bind prothrombin or factor X or to present negatively charged phospholipids on their surface. They may, however, contain NAD(P)H oxidase activity, which has the potential to generate reactive oxygen species that contribute to endothelial cell apoptosis in sepsis.491
An intricate pathway of protein–protein interactions has been proposed for platelet secretion in which granules tether and dock to the inner leaflet of the plasma membrane, after which fusion of the two opposing lipid bilayers mediates cargo release.492 Docking and tethering are thought to be, in part, mediated by small GTP-binding proteins of the Rab family. Platelets have been reported to contain at least 11 Rabs, although only a few have been shown to be functionally relevant. Rab27s a and b are important for both granule biogenesis and secretion,493 whereas Rab 4 appears to have a role in secretion.494 The α-granule–associated Rab6 was shown to be phosphorylated upon thrombin stimulation in a protein kinase C (PKC)-dependent manner, and phosphorylation seems to increase its GTP loading.495
Platelet granule–plasma membrane fusion is analogous to exocytosis in neurons, where detailed studies have shown the importance of a core set of integral membrane proteins called soluble N-ethylmaleimide-sensitive factor (NSF) attachment protein receptors (SNAREs).496 It is generally accepted that vesicle/granule-target membrane fusion is governed by the binding of a SNARE from the cargo-containing granule or vesicle (v-SNARE) with a heteromeric protein complex in the target membrane (t-SNAREs). The resulting, trans-bilayer complex is minimally sufficient for membrane fusion.497 In human platelets, the v-SNAREs are vesicle-associated membrane protein (VAMP)-2/synaptobrevin, VAMP-3/cellubrevin, VAMP-7/TI-VAMP, and VAMP-8/endobrevin, with the latter being most abundant.498–502 There are two classes of t-SNAREs: the synaptosome-associated protein (SNAP)-23/25/29 type and the syntaxin type. Human platelets contain syntaxins 2, 4, 7, and 11498–502 as well as SNAP-23, -25, and -29.503,504 Functional studies using in vitro assays and genetically engineered mice have established that VAMP-8 is the primary v-SNARE required for secretion from all three classes of platelet granules.501,502 VAMP-2 or VAMP-3 can also play a role at higher levels of stimulation. As for t-SNAREs, SNAP-23 and syntaxin 2 are required for each secretion event. Syntaxin 4 appears to also play a role, but only in α-granule and lysosome release.505–508
Although the SNARE proteins are sufficient to mediate membrane fusion, they do so inefficiently and thus require accessory proteins to control where and when they interact. Many of these regulators may be sensitive to second messengers such as diacylglycerol (DAG) and Ca2+, whereas others are substrates for kinases, such as PKC. The Munc18 family (a, b, and c) controls syntaxins and is critical for platelet secretion.509–511 Studies show that Munc18a and c are phosphorylated by PKC upon platelet activation and that this affects Munc18/syntaxin binding affinity.510,511 At least two members of the Munc13 family are present in platelets (Munc13–1 and Munc13–4) (Schraw TD, Ren Q, and Whiteheart SW, unpublished data).512 Munc13–4 appears to be important for dense granule release and functions through its interactions with Rab27.513,514 Munc13s have Ca2+ and DAG binding sites and thus may be regulated by the secondary messengers generated during platelet activation.
Munc13–4 has drawn particular attention based on its involvement in familial hemophagocytic lymphohistiocytosis (FHL) and Griscelli syndrome. Munc13–4 is mutated in type 3 FHL515 and interacts with the protein mutated in type 2 Griscelli syndrome, namely Rab27a.516 One feature common to both diseases is the inability of T cells to properly organize the cytotoxic synapse required for toxin secretion and target cell killing.515 For FHL patients, it is not clear whether they have bleeding-time defects because they generally receive marrow transplants very early in life.
Platelet granule–plasma membrane fusion is mechanistically analogous to exocytosis in neurons and other secretory cell types, where detailed studies have demonstrated the importance of a core set of integral membrane proteins called SNAREs.517,518 It is generally accepted that vesicle/granule-target membrane fusion and, thus, granule content release require the binding of a SNARE from the cargo-containing granule or v-SNARE, with a heteromeric protein complex in the t-SNAREs. The resulting, trans-bilayer complex is minimally sufficient for membrane fusion.519 In human platelets, the detectable v-SNAREs are VAMP-2/synaptobrevin, VAMP-3/cellubrevin, VAMP-4, VAMP-5, VAMP-7/TI-VAMP, and VAMP-8/endobrevin, with VAMP-8 being most abundant.501,502,520–523 There are two classes of t-SNAREs: the SNAP-23/25/29 type and the syntaxin type. Human platelets contain syntaxins 2, 4, 6, 7, 8, 11, 12, 16, 17, and 18.501,502,520–524 SNAP-23, -25, and -29 are all detectable, but SNAP-23 is the most abundant.521,525,526 Functional studies, using in vitro assays and genetically altered mice, established that VAMP-8 is the primary v-SNARE required for secretion from all three classes of platelet granules; however, platelets lacking VAMP-8 do release their contents at attenuated rates, suggesting roles for other VAMPs.502 Differential usage of the VAMPs may allow platelets to fine tune their release of cargo. For t-SNAREs, patients with FHL4, who are deficient in syntaxin 11, show robust platelet secretion defects from all three granule types.527 Studies of mouse platelets suggest a minor role for syntaxin 8,524 but loss of syntaxin 2 and/or syntaxin 4 had no effect.527 Syntaxins form a heterodimeric complex with SNAP-23/25–like t-SNAREs. In platelets, SNAP-23 is the critical family member, based on its abundance and results from in vitro assays.505,506,528 SNAP-23 phosphorylation, by IκB kinase (IKK), is important for SNARE complex assembly, membrane fusion, and secretion. Platelet-specific loss of IKK or its pharmacologic inhibition leads to bleeding.529
Although the SNARE proteins mediate membrane fusion, they do so inefficiently and thus require accessory proteins to control where, when, and how they interact. Many of these regulators are sensitive to second messengers (e.g., calcium). The Sec1/Munc18 (SM) proteins are syntaxin chaperones that control how the t-SNAREs interact with other SNAREs.510,530–533 Although several isoforms are present, only Munc18b is important for platelet exocytosis.530,532,534 It chaperones syntaxin 11 and is defective in FHL5 patients. Other SM proteins (e.g., Vps33a/b) are important for granule biogenesis.535,536 Another syntaxin regulator, tomosyn-1/syntaxin binding protein 5 (STXBP5), binds to syntaxin/SNAP-23 heterodimers and affects access to v-SNAREs.537 Genome-wide association studies (GWAS) suggest that alterations in STXBP5 are linked to venous thrombosis risk resulting from increased plasma VWF.537–539 Surprisingly, mice lacking STXBP5 have a severe arterial bleeding diathesis as a result of their defective platelet secretion.537,539
Munc13 family members contain binding sites for calcium, phosphatidylserine, DAG, and calmodulin.540 Two major family members, Munc13–2 and Munc13–4, are detectable in platelets, although only Munc13–4 is functionally relevant.521,541 Munc13s are generally thought to be docking factors that localize granules for membrane fusion. Both FHL3 patients and the Unc13dJinx mouse strain lack Munc13–4 and have robust granule-release defects and bleeding diatheses.541,542 Munc13–4 binds to a small GTP-binding protein called Rab27, which is also important for platelet exocytosis and is defective in Griscelli syndrome.543 Another Rab27-binding protein, called synaptotagmin-like protein 4/granuphilin, is also reported to be important for platelet exocytosis.544
Three types of FHL are caused by defects in genes encoding proteins that are important in platelet secretion: Munc13–4/FHL3, syntaxin 11/FHL4, and Munc18b/FHL5. Rab27a is defective in a related disease, Griscelli syndrome. One feature common to both diseases is the inability of T cells to properly organize the cytotoxic synapse required for toxin secretion and target cell killing. This suggests that these T-cell populations and platelets share common secretory machinery elements. For FHL patients, it is unclear whether bleeding or defective platelet function can be used as diagnostic criteria, but they have been reported as symptoms.545
The Homo sapiens genome is comprised of approximately 3.2 billion base pairs and has approximately 3.5 million single nucleotide polymorphisms (SNPs) that occur at frequencies of 1 percent or greater, but continued sequencing of more genomes indicates there are at least an additional 43 million rare or “private” SNPs. dbSNP (www.ncbi.nlm.nih.gov/projects/SNP) maintains information about sequence variation, allele frequencies, differences in frequencies between populations of different ethnicity, and their functional consequences. New technologies, such as next-generation sequencing, and new analytic methodologies have driven and continue to drive expansion of genomics at a very rapid pace. Both epidemiologic and experimental approaches have been and are used to assess significance and functionality of platelet gene variants and gene expression, including genetic epidemiology, biochemistry, cell biology, physiology, and animal studies.
Because platelets play a central role in acute ischemic syndromes, antiplatelet therapy is a mainstay of therapy. Platelets may also contribute to the pathophysiology of the chronic process of atherosclerosis,546 although the data are less consistent than with thrombosis. The earliest platelet genetic association studies considered associations between atherothrombotic disease outcomes and candidate platelet gene variants that altered amino acids in platelet membrane adhesion receptors. Numerous studies reported associations between myocardial infarction and stroke with SNPs in integrin β3 (ITGB3), integrin α2 (ITGA2), and GPIbα (GP1BA), and GPVI (GP6).547 In these relatively small studies, positive associations were more likely to be observed in patients with acute thrombosis and less likely in patients with stable atherosclerosis. However, there were inconsistent and conflicting results in these candidate studies.
No unbiased GWAS has been performed using documented arterial thrombosis as the clinical phenotype, but many have been performed with coronary artery disease (CAD). Multiple studies have demonstrated that the Chr9p21.3 locus is associated with both myocardial infarction (MI) and CAD. A meta-analysis of all CAD GWAS studies that included 63,746 patients with acute and chronic CAD and 130,681 controls identified 46 loci meeting genome-wide significance.548 These loci explain less than 11 percent of CAD heritability, and although a substantial proportion of the identified genes regulate lipid metabolism and inflammation, most loci are not located in previously well-known or well-characterized platelet genes. Few of these loci have been tested for functional effects in platelets. There are numerous possible explanations for the nonassociation with well-studied platelet candidate genes, including small platelet gene effect sizes in underpowered heterogeneous clinical phenotypes.549
Whole-exome sequencing is well suited to identify variants in protein-coding genes and was used to identify NBEAL2 as the causative gene in the gray platelet syndrome.550,551
The CYP2C19 * 2 allele (681G>A; rs4244285) causes a loss of function of the CYP2C19 enzyme and reduced platelet inhibition by clopidogrel as a result of decreased conversion to the active metabolite.552 It could account for approximately 12 percent of the variation in platelet inhibition in response to clopidogrel.553 Several large meta-analyses have shown the loss-of-function CYP2C19 * 2 allele is associated with both stent thrombosis and cardiovascular ischemic events or death in patients undergoing percutaneous coronary interventions.554,555
Many cellular pathways in multiple tissues contribute to the pathogenic processes resulting in an atherothrombotic event (Fig. 2–7), increasing the “noise” in genetic epidemiology studies testing for associations with complex phenotypes. The use of intermediate phenotypes as outcomes in genetic association studies has enhanced power to detect gene associations because the number of genes potentially responsible for the phenotype is reduced, thereby increasing the fraction of the variance explained by any single factor or gene. Despite large interindividual variability in platelet reactivity, light transmission aggregometry has been shown to be reproducible and heritable, with the reproducibility persisting for years.556,557
Figure 2–7.
Intermediate phenotypes in association studies. Atherothrombosis is a complex phenotype that is regulated by many intermediate traits, of which platelet reactivity is only one. Because a large number of genes contribute to multiple traits, the effect of any one gene on atherothrombotic events, such as myocardial infarction, is small. This highly simplified diagram assumes five traits each contribute 20 percent to the complex trait (heavy solid arrows) and two different genes equally regulate each intermediate trait. Thus, each gene contributes 50 percent to the intermediate trait (thin arrows), but only 10 percent to the clinical end point (faint dashed arrows). Thus, for any given sample size, there is more power to detect genetic associations with intermediate phenotypes than with complex traits. (Reproduced with permission from Bray PF: Platelet genomics beats the catch-22, Blood. 2009 Aug 13;114(7):1286–1287.)
The Leu33Pro variant of integrin β3 (rs5918 of ITGB3) is responsible for human platelet alloantigen 1a/b (PlA1/PlA2).558 Fibrinogen and prothrombin binding is enhanced to the Pro33 isoform of purified integrin αIIbβ3.559 Compared to cell lines expressing Leu33 variant of integrin, Pro33 cells have increased adhesion, spreading, actin cytoskeletal reorganization, and migration under static560,561 and shear conditions.562 This prothrombotic phenotype of Pro33 is mediated by enhanced outside-in platelet signaling through integrin αIIbβ3.563,564 Notably, this variant does not affect inside-out signaling, as assessed by standard platelet light transmission aggregometry of human platelets.565 Additional support for the prothrombotic nature of the Pro33 variant of integrin β3 comes from mice made homozygous for Pro33. These animals have reduced bleeding, increased in vivo thrombosis and enhanced outside-in integrin αIIbβ3 signaling, but normal inside-out signaling.566
Laboratory evidence for functional effects of genetic variants in the gene encoding GPIbβ has been inconsistent. Variants in the two platelet collagen receptors, GPVI and integrin α2 subunit (of integrin α2β1) alter receptor expression and adhesion to collagen using in vitro perfusion assays.567–569 Functional variants in the genes encoding FcγRIIA (FCGR2A), P2Y12 (P2RY12), GPIV (CD36), and PAR-1 (F2R) have also been reported.
Associations between SNPs in 97 hematopoietic cell genes were tested, and 17 novel associations with platelet responses to crosslinked collagen-related peptide (CRP) and ADP were identified, including genes encoding cell surface receptors (CD36, GP6, ITGA2, PEAR1, and P2Y12), kinases (JAK2, MAP2K2, MAP2K4, and MAPK14), and other signaling molecules (GNAZ, VAV3, ITPR1, and FCERG1).570 Variants at the Chr9p21.3 locus are associated with the platelet aggregation response to low (0.5 mcg/mL) but not higher concentrations of collagen in a large cohort with two replication studies.571
The first GWAS reported for platelet reactivity tested association of 2.5 million SNPs with platelet aggregation responses to ADP, collagen, and epinephrine.565 The primary cohorts were generally healthy, European-ancestry populations from the Framingham Heart Study (FHS) (n = 2753) and the GeneSTAR cohorts (n = 1238). SNPs at seven loci (PEAR1, MRVI1, SHH, ADRA2A, PIK3CG, JMJD1C, and GP6) met genome-wide statistical significance and were replicated in an African-ancestry cohort (n = 840). A second platelet function GWAS identified SNPs in SVIL (encodes supervillin) as associated with closure time in the in vitro platelet function analyzer PFA-100.572 Human platelet gene expression studies and data with Svil –/– mice demonstrated an inhibitory role for supervillin in platelet adhesion and thrombus formation under high-shear but not low-shear conditions. A meta-analysis by the HaemGen consortium of 66,867 individuals identified 43 and 25 loci associated with platelet number and mean platelet volume (MPV), respectively.573 These loci accounted for 4.8 percent of the phenotypic variance in platelet number and 9.9 percent in MPV and included well-known platelet regulators (ITGA2B, GP1BA, and F2R). These investigators identified 11 of the genes as novel regulators of blood cell formation using gene silencing in Danio rerio and Drosophila melanogaster.
The Homo sapiens genome includes approximately 21,000 protein-coding genes (genome build GRCh38). To date, more than 10 times this number of protein-coding transcripts have been identified, primarily as a result of alternate exon splicing, and more are being continually discovered. Platelets from healthy subjects contain approximately 2.20 femtograms (fg) of total RNA per cell, which is approximately 1000-fold less than nucleated blood cells. Platelets can splice pre-mRNA into mature mRNA, which is translated into proteins.574,575 Characterization of the transcriptome enables quantitative assessment of gene expression in the tissue of interest and identification of alternately spliced transcripts. Genome-wide transcriptome studies have enabled dissection of the molecular basis of inherited platelet disorders and a better understanding of the relationship between gene expression and megakaryocyte and platelet differentiation. In addition, platelet RNA profiles may have utility as biomarkers.576
Technologic advances have greatly facilitated understanding the platelet transcriptome. Early studies using serial analysis of gene expression and microarrays estimated approximately 6000 mRNAs in the human platelet.577,578 Platelet RNA sequencing (RNA-seq) has demonstrated an unexpected complexity to the transcriptome and substantive differences between the human and mouse platelet transcriptome.579 The exquisite sensitivity of RNA-seq provided estimates of approximately 9000 protein-coding genes in platelets (Fig. 2–8),580,581 although only approximately 7800 are commonly expressed582 in human platelets. Approximately half of the transcripts in platelets encode mitochondrial genes.581 Platelet mitochondrial mRNAs are inversely correlated with subject age,582 and mitochondrial function may regulate platelet apoptosis583 and support optimal platelet function during storage,584 but platelet mitochondria diseases have not been described. The S-shaped curves in Fig. 2–8 illustrate several features of the human platelet transcriptome: (1) estimates of expressed protein-coding genes are more similar among different subjects for high-abundance genes (leftward in Fig. 2–8), and (2) there is substantial interindividual variation in total transcript estimates when considering the less abundant genes (rightward in Fig. 2–9). Furthermore, it is not known what is the biologically relevant copy number of transcripts in any cell, and the arbitrary choice of “threshold” could dramatically affect the number of reported genes expressed in platelets. Transcriptomes of primary megakaryocytes have not been determined, but RNA profiling of megakaryocytes derived from cultured CD34+ hematopoietic stem cells has identified transcripts that are differentially expressed upon differentiation and between normal subjects and patients with essential thrombocytosis.585,586
Figure 2–8.
Estimates of platelet-expressed mRNAs. Platelet total RNA was extracted from four normal donors, depleted of ribosomal RNA (rRNA), and subjected to RNA sequencing (RNA-seq). The number of platelet-expressed mRNAs (y axis) was plotted against RNA-seq read number in log2 ratios normalized to β-actin. (Reproduced with permission from Bray PF, et al: The complex transcriptional landscape of the anucleate human platelet, BMC Genomics 2013 Jan 16;14:1)
Figure 2–9.
Platelet–leukocyte interactions. A number of interactions can occur between platelets and leukocytes, including neutrophils and monocytes. The interaction between platelet P-selectin and leukocyte P-selectin glycoprotein ligand-1 (PSGL-1) probably is the most important initial interaction (and can lead to tissue factor synthesis by monocytes), but fibrinogen binding simultaneously to activated αMβ2 on leukocytes and either αIIbβ3 or αVβ3 on platelets may play a role under certain circumstances. Platelets can release platelet-activating factor (PAF), which can interact with a PAF receptor (PAFR) on leukocytes, leading to αMβ2 activation and binding of fibrinogen and factor X. Leukocyte αMβ2 can also interact with platelet junctional adhesion molecule-3 (JAM-3) or GPIb. Platelets can release chemokines (e.g., ENA-78, GRO-α, and RANTES [regulated upon activation, normal T-cell expressed and secreted]), and β-thromboglobulin (βTG) released by platelets can be converted by leukocyte cathepsin G (CG) into the potent chemotactic CXC chemokine NAP-2. Some of the chemokines, in turn, activate leukocytes by binding to the chemokine receptor CXCR2. Platelets also contain the potent immune-stimulating molecule CD40 ligand (CD40L), and both express it on the platelet surface and release it into the circulation upon platelet activation. The interaction between thrombospondin and CD36 molecules on both platelets and some leukocytes and the presence of CD40 on platelets are not shown. VWF, von Willebrand factor.
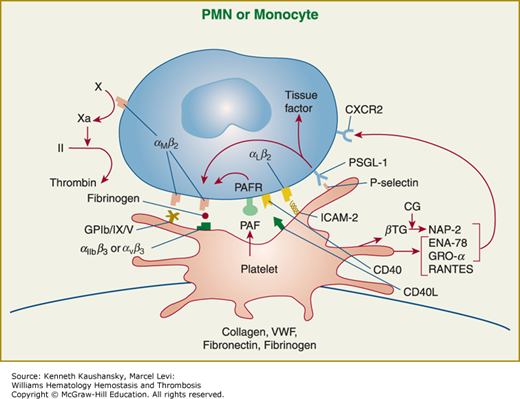
Platelet mRNA profiling in patients with acute ST-segment elevation MI and stable CAD demonstrated that S100A9 (myeloid-related protein-14 [MRP-14]) was expressed at higher levels in patients than controls.587 This discovery was validated in the Women’s Health Study and PROVE IT-TIMI 22 trials.587,588 Platelet mRNA expression profiling can distinguish essential thrombocythemia (ET) patients from healthy subjects,589 and levels of HIST1H1A, SRP72, C20orf103, and CRYM can predict JAK2 V617F–negative ET in 87 percent of patients.590 mRNA expression profiling identified reduced MYL9 transcripts in platelets of a patient with an inherited platelet defect.591 Platelet RNA-seq was also used to identify NBEAL2 as causing the gray platelet syndrome.592
An unbiased genome-wide platelet RNA expression study identified an association between expression of PEAR1 and platelet activation.593 A similar approach identified 290 differentially expressed transcripts between hyperreactive versus hyporeactive platelets.594 mRNA and protein levels of VAMP-8, a critical v-SNARE involved in platelet granule secretion, were significantly higher in hyperreactive platelets. Another study identified 63 genes differentially expressed according to platelet activation by ADP and/or CRP.595 Two of these genes, COMMD7 and LRRFIP1, were associated with early-onset MI.595 The Platelet RNA and Expression-1 (PRAX1) study phenotyped platelet function and performed genome-wide platelet RNA expression profiling on 70 black and 84 white subjects.596 PAR4-mediated platelet aggregation and calcium mobilization were greater in black subjects than white subjects. A novel platelet gene encoding phosphatidylcholine transfer protein (PC-TP) showed a strong correlation with race and with PAR-4 reactivity, and a PC-TP–specific inhibitor blocked PAR-4– but not PAR-1–mediated platelet aggregation. This finding underscores the genetic basis for interindividual variation in platelet function and the potential need to consider race and genetic factors when treating patients with antiplatelet therapies.
The best studied of the noncoding RNAs are microRNAs (miRNAs), which regulate expression of more than 60 percent of protein-coding genes.597,598 Human platelets express approximately 200 annotated miRNAs, some of which are differentially expressed according to platelet reactivity and may predict platelet responsiveness to activation599 and some of which are differentially expressed by age, gender, and race.582,596 Indirect evidence indicates strong correlations between megakaryocyte and platelet miRNA levels.600 miR-155 maintains megakaryocyte progenitors in an undifferentiated state,601 whereas miR-150 and miR-125b-2 drive megakaryocyte differentiation.602,603 Loss of expression of miR-145 in the 5q– syndrome leads to an increase in the megakaryocyte Fli-1 transcription factor, thus enhancing megakaryocyte production.604
Platelet miRNA profiles are more stable than mRNA profiles and are useful as biomarkers.576 Levels of miR-26b and miR-28 are associated with myeloproliferative neoplasms,605,606 whereas levels of miR-10a, miR-148a, and miR-490–5p discriminate ET from secondary thrombocytosis.607 Specific sets of platelet miRNAs have been associated with MI.608,609 Antiplatelet therapies alter platelet miRNA levels.610,611 Relationships between platelet miRNAs, mRNAs, and physiology in the same subjects permit prediction of miRNA function and discovery of novel platelet genes.599 This approach identified PRKAR2B as associated with platelet reactivity, and a functional effect was confirmed in murine platelets lacking Prkar2b.599 A similar approach was used to demonstrate that platelet miR-376c levels were higher in white subjects compared to black subjects and that these levels correlated with PCTP mRNA, PC-TP protein, and platelet PAR-4 reactivity.596 miR-376c directly targets the PCTP 3′UTR and represses its expression.596
Disease pathophysiology is dictated by the effects of proteins, including their levels, structures, and posttranslational modifications. Cataloging platelet proteomes in health and disease and under different activation states provides information not achievable from genomics or transcriptomics, including protein isoforms, localization, stoichiometry, and posttranslational modifications. Early proteome-wide studies of platelet lysates used two-dimensional gel electrophoresis (2D-GE).612 However, technologic advances using nongel approaches with proteolytic peptide analyses have largely replaced 2D-GE and include surface-enhanced laser desorption/ionization (SELDI), isotope-coded affinity tags (iCAT), and isotope tags for relative and absolute quantification (iTRAQ).613,614 These improved technologies have provided an estimate of approximately 20 million protein molecules per platelet and have updated estimates of the number of detectable different proteins in the platelet proteome to nearly 5000.615 Pathway and gene ontology analyses reveal most highly expressed platelet proteins localize to the cytoplasm, with substantial percentages in the membrane or secretome,616 and fall into expected functional categories of cytoskeletal rearrangement, membrane trafficking, and intracellular signal transduction.615
Platelet protein levels are regulated by mRNA translation in megakaryocytes and platelets, uptake of plasma proteins, and protein degradation,574,617 although the relative contribution of each mechanism to the platelet proteome in health and disease is unknown. The dynamic nature of the platelet proteome is illustrated by alterations with disease, aging, gender, and other environmental factors,616 as well as differential sorting of proteins between megakaryocytes and platelets.618 Infectious agents, such as dengue virus, stimulate blood platelet mRNA translation into protein.619 Posttranslational modifications of platelet proteins, such as phosphorylation, have critical effects on platelet activation. Platelets from healthy individuals exhibit marked interindividual variation in function,556 and unbiased genome-wide approaches have identified variation in proteins regulating the corresponding function.594 Components of protein ubiquitination and degradation have been identified in platelets, but their function is poorly understood.
Most platelet proteomic analyses to date have studied platelets from small numbers of healthy donors. Analyses of resting whole platelets have provided global protein profiles.612,620 Fractionation of platelet lysates has been used to assess the α granule,621 dense granule,622 and membrane proteomes.623,624 Proteins with posttranslational modifications have been identified for phosphorylation,625,626 palmitoylation,627 and glycosylation.628 After platelet activation, hundreds of proteins have been identified in releasates (secretomes)629,630 and microparticles.631,632
Platelet proteome-wide analyses were used to identify NBEAL2 as the gene responsible for the gray platelet syndrome551 and to unravel the molecular basis of the Quebec platelet disorder.633 Differentially expressed platelet proteins involved in integrin αIIbβ3 signaling were observed in the myelodysplastic syndrome.634 Proteomic approaches have consistently identified platelet septin and actin as increasing over time in storage.635–637 A small study suggested platelet protein posttranslational modifications may be associated with acute coronary syndromes.638
Transcriptomic approaches have identified about twice as many genes expressed in platelets as have proteomic approaches, primarily because the former has greater sensitivity. Correlations between 10 platelet RNA-seqs and the most quantitatively robust proteomic analyses to date have been reported.639 Most (87.8 percent) proteins had a detectable corresponding mRNA, and the relative abundances showed a significantly positive, albeit weak, correlation. Platelet proteins that lack a corresponding mRNA are likely to be taken up from plasma rather than being synthesized in megakaryocytes, and include fibrinogen, albumin, and immunoglobulins, all of which were suspected to fall into this category based on other studies.640 Platelet mRNAs that lack a corresponding protein may be vestigial from the megakaryocyte. Some of these could be translated subsequently by the platelet under physiologic demands. Combining “multiomic” data with phenotyping can provide important insights as demonstrated by a study in which transcriptomic and proteomic analysis identified six platelet transcripts associated with aspirin resistance.641 The expression of these genes was associated with death or MI. In addition, platelet phenotyping and genome-wide genotyping and platelet mRNA and miRNA profiling led to the identification of novel protein-coding and noncoding transcripts associated with platelet activation.596
In resting platelets, negatively charged phospholipids, including phosphatidyl serine (PS) and phosphatidylethanolamine (PE), are almost exclusively present in the inner leaflet of the cell membrane and phosphatidylcholine predominates in the outer leaflet. This asymmetry is maintained by ATP-dependent “flippase” transporters, which restrict PS to the inner membrane surface, and “floppases,” which promote outward-directed lipid transport.84,85,642,643 When platelets are activated by strong agonists, negatively charged phospholipids redistribute to the outer leaflet of the platelet plasma membrane. This involves a putative calcium-dependent “scramblase” that transports lipids bidirectionally and, when active, collapses membrane asymmetry and results in PS exposure on the outer leaflet. The eight-transmembrane domain containing protein TMEM16F serves as a Ca2+-activated, nonselective cation channel that is crucial for Ca2+-dependent phospholipid scrambling and PS exposure on activated platelets.644
Platelet activation with strong agonists also results in the formation of microparticles, which are particularly rich in surface-exposed negatively charged phospholipids. Microparticles also are rich in factor Va and thus actively support thrombin generation.82,645,646 Microparticle formation can be induced in vitro by activation of platelets with ionophore A23187, complement C5b-9, or the combination of thrombin and collagen; by adding tissue factor to recalcified platelet-rich plasma; or by high shear stress.645,647–652 Elevations of cytosolic Ca2+, calpain activation, cytoskeletal reorganization, protein phosphorylation, and phospholipid translocation have all been implicated in microparticle formation.
The biologic relevance of platelet microparticles is supported by the finding of increased circulating levels of platelet microparticles in patients with activated coagulation and fibrinolysis, diabetes mellitus, sickle cell anemia, human immunodeficiency virus infection, unstable angina, heparin-induced thrombocytopenia with thrombosis, and respiratory distress syndrome.645,653 Microparticles can bind to fibrin thrombi via one or more of the receptors present on their surface, including integrin αIIbβ3, GPIb/IX, P-selectin, and possibly P-selectin glycoprotein ligand (PSGL)-1.654
Microparticles bind factors VIII, Va, and Xa, allowing them to form both the factor Xase and prothrombinase complexes on their surface.645 They can also bind protein S and facilitate inactivation of factors Va and VIIIa, which could serve an anticoagulant function.655,656 In addition, microparticles can activate platelets by supplying arachidonic acid.
Evidence supporting the importance of platelet microparticle formation to platelet coagulant activity has been gathered from observations of patients who have significant bleeding diatheses in association with defects in platelet microparticle formation (Scott syndrome; Chap. 11).657–659 Platelets from the most intensively studied patient had an impaired ability to accelerate the activation of both factor X and prothrombin. In addition, this patient’s platelets exhibited both abnormal factor V binding and abnormal exposure of negatively charged phospholipids.
Activated platelets synthesize tissue factor by splicing pre-mRNA into mature mRNA and then translating the tissue factor protein.660,661 Additionally, platelet thrombi can recruit tissue factor from blood by binding leukocyte-derived, tissue factor-containing microparticles or by binding an alternatively spliced, soluble form of tissue factor.466,472,662–665 The interaction between PSGL-1 on the surface of leukocyte-derived microparticles and P-selectin on the surface of activated platelets appears to play an important role in the binding of microparticles to platelet thrombi.664 Interactions between platelets and leukocytes, and perhaps leukocyte-derived microparticles, reportedly enhance (“de-encrypt” or decrypt) tissue factor activity, probably by supplying negatively charged phopholipids666 and/or the oxidoreductase enzyme protein disulfide isomerase (PDI).667
Platelet-dense granules contain polyphosphate, a linear polymer of inorganic phosphate synthesized by inositol hexakisphosphate 6 kinase. Polyphosphates are released during platelet activation and promote clot formation. Polyphosphates affect many steps in coagulation. Polyphosphates accelerate factor V and factor XII668 and alter the structure of fibrin clots. In the presence of polyphosphates, fibrin clots have thicker fibers and are more resistant to fibrinolysis.669 In contrast to bacterial polyphosphates, which are long-chain structures, platelet polyphosphates have shorter chain length and are more effective in increasing factor V and TFPI activity.
Incontrovertible evidence exists that platelets accelerate thrombin formation.658,659,670–672 Platelets accelerate the activation of factor X by factors IXa and VIIIa and the activation of prothrombin by factors Xa and Va.659,670 However, only a subpopulation of platelets develops a procoagulant phenotype with activation, as only a fraction of activated platelets display high levels of factors Va and Xa, termed “coat” platelets.464,465,670,673 The assembly of the factor IXa/factor VIIIa/platelet complex increases the catalytic efficiency of factor X activation (kcat/Km [turnover number/Michaelis-Menten dissociation constant]) by a factor of 2.4 × 106.670 Prothrombin binds to approximately 20,000 sites on activated platelets with a KD equal to its plasma concentration (approximately 0.15 μM).674 Integrin αIIbβ3 binds prothrombin through its RGD domain and may contribute to the localization of prothrombin to the surface of unactivated and activated platelets.675
In addition to accelerating coagulation, the binding of activated coagulation factors to the surface of platelets appears to protect them from inactivation by inhibitors in plasma and platelets.399 The bleeding diathesis in patients with Quebec platelet syndrome, who have proteolysis of their platelet α-granule factor V, supports the potential importance of platelet factor V in normal hemostasis (Chap. 11), as do the studies of another patient with abnormal platelet factor V.659
Other connections between platelets and the coagulation system include: (1) the presence of fibrinogen in platelet α granules and perhaps on the surface of platelets, where it is strategically located for interactions with locally generated thrombin371,399; (2) the presence of intracellular VWF and the binding of extracellular VWF to platelets (via GPIb/X and integrin αIIbβ3), with the potential colocalization of factor VIII attached to the VWF (Chap. 16); (3) activation of factor XI by thrombin on the platelet surface,676,677 with the dimeric structure of factor XI allowing it to interact both with the platelet and factor IX simultaneously678; (4) a factor XI-like protein associated with platelet membranes, which may be an alternatively spliced form of factor XI lacking exon V; the level of this factor appears to correlate better with hemorrhagic symptoms than does the level of plasma factor XI399,679; (5) the presence of cytoplasmic factor XIII (Chap. 3); (6) the presence of inhibitors of coagulation (α1-protease inhibitor, C-1 inhibitor, TFPI, the thrombin inhibitor protease nexin I, and the factors IXa and XIa inhibitor protease nexin II or β-APP)399,448; and (7) promotion of factor XII activation by ADP-treated platelets.399
The interactions between platelets and the fibrinolytic system are complex; Table 2–3 contains a partial listing of reported findings.680–684 Both profibrinolytic398,685–692 and antifibrinolytic693–701 effects of platelets have been described, and so it is difficult to predict the net effect. Since platelet-rich thrombi are known to resist thrombolysis in animal models, the antifibrinolytic effects of platelets appear to predominate in vivo.702
| Profibrinolytic effects of platelets |
| Tissue plasminogen activator (t-PA) and single-chain urokinase-type t-PA identified on or in platelets. |
| Unactivated platelets bind plasminogen, and binding is enhanced by thrombin. |
| Thrombospondin, a plasminogen-binding protein, is expressed on the surface of platelets after activation. |
| Activation of plasminogen by t-PA is enhanced by platelets. |
| Clot lysis is enhanced by platelets in some model systems. |
| Antifibrinolytic effects of platelets |
| Plasminogen activator inhibitor-1 and α2-antiplasmin are present in platelet granules. |
| Platelets contain protease nexin-1, a serpin that inhibits plasminogen activators and plasmin. |
| Platelets contain factor XIII, which can crosslink fibrin, making it resist fibrinolysis, and can crosslink α2-antiplasmin to fibrin, enhancing its antifibrinolytic effects. |
| Platelets contain tissue factor pathway inhibitor-2, which inhibits t-PA. |
| Platelet αIIbβ3 can bind plasma factor XIIIa directly or indirectly, localizing it to the site of thrombus formation. |
| Platelets facilitate clot retraction, which diminishes the efficiency of fibrinolysis. |
| Platelet-activating effects of thrombolytic agents |
| Streptokinase and t-PA activate platelets in vivo and in vitro. |
| Plasmin, at high doses, can aggregate platelets. |
| Thrombolytic agents may paradoxically generate the potent platelet agonist thrombin or release it from thrombi. |
| Thrombolytic agents may blunt the prostacyclin increase that accompanies acute thrombosis. |
| Platelet-inhibiting effects of thrombolytic agents |
| Plasmin, at low doses, can inhibit platelet activation and aggregation. |
| Platelets can be disaggregated by t-PA by selective lysis of platelet-bound fibrinogen. |
| Plasmin can cause redistribution and/or cleavage of platelet glycoprotein Ib. |
| Inhibition of platelet aggregation by the depletion of plasma fibrinogen, if severe, and generation of fibrin (ogen) degradation products. |
| Proteolysis of plasma von Willebrand factor. |
| Prolongation of the bleeding time. |
The effects of fibrinolytic agents on platelets are similarly complex. For example, there is considerable evidence that fibrinolytic agents can activate platelets soon after administration,703–709 via either a direct effect of plasmin,710–713 perhaps acting on PAR-4,714 or an indirect effect through the paradoxical generation of thrombin.683,715–718 Interpretation of the latter studies is complicated by the ability of tissue plasminogen activator to release fibrinopeptides from fibrinogen, one of the biomarkers used to assess thrombin activation.719
Stimulation of platelets by thrombolytic agents may prolong the time required for reperfusion of thrombosed blood vessels and may contribute to reocclusion after successful reperfusion.680,720 In animal models and in humans, potent antiplatelet agents can, in fact, speed reperfusion, abolish reocclusion, and diminish the size of myocardial infarcts.721–723 In human studies, the benefits of combining integrin αIIbβ3 antagonists with fibrinolytic agents in enhancing coronary thrombolysis have been counterbalanced by an increase in major hemorrhage.724 Combining a potent integrin αIIbβ3 antagonist with a reduced dose of a fibrinolytic agent in acute ST-segment elevation MI when patients are rapidly treated with percutaneous coronary intervention has demonstrated evidence for more rapid reperfusion, but clinical benefit has been variable and bleeding has been increased.725,726 In experimental models of stroke, paradoxically, early treatment with integrin αIIbβ3 antagonists reduces the hemorrhage associated with thrombolytic therapy, perhaps by preventing platelet aggregation in the microcirculation and the release of agents that can damage the vasculature and diminish its integrity.727–729 In human studies, however, a potent integrin αIIbβ3 antagonist given alone did not improve clinical outcomes.730,731
With prolonged use of thrombolytic agents, inhibition of platelet function can occur via a variety of mechanisms.102,707,708,732–744 These effects may contribute to some of the hemorrhagic phenomena observed with this therapy. One proposed mechanism is that the thrombolytic agents make platelets refractory to further stimulation by agonists.
Leukocytes can bind to activated platelets and in model systems transmigrate through a platelet monolayer (reviewed in Ref. 745; see Fig. 2–9). Animal models and studies of human tissue demonstrate that within hours after vascular injury, leukocytes become enmeshed in platelet thrombi and/or transiently form a monolayer on top of adherent or aggregated platelets.746,747 These interactions may be important at sites of vascular injury or inflammation where leukocytes have been shown to deposit on adherent and aggregated platelets. Platelet recruitment of leukocytes has been associated with a number of systemic and inflammatory processes in animal models, including the development of intimal hyperplasia after vascular injury,748 ischemia–reperfusion injury, alloimmunity-mediated transplant rejection,749 obesity,750 and acute lung injury.751 By depositing chemokines such as CCL5 (also termed RANTES [regulated upon activation, normal T-cell expressed and secreted]) on activated endothelium752,753 or by direct interactions with leukocytes,754 platelets may also enhance leukocyte recruitment to inflamed or atherosclerotic endothelium and thereby promote the development and progression of atherosclerosis.
Many mechanisms of platelet–leukocyte interactions have been defined, but the initial interaction appears to be mediated primarily by the interaction between P-selectin (CD62P) expressed on the surface of activated platelets and PSGL-1 on the surface of neutrophils and monocytes.755–761 P-selectin–PSGL-1 interactions are characterized by rapid on-and-off rates that promote tethering and rolling of leukocytes along adherent platelets. In addition to PSGL-1, leukocyte CD24 may also bind P-selectin. The transient P-selectin–mediated interactions are stabilized by subsequent contacts mediated, in large part, by activation of leukocyte β2 integrins. Platelet surface-immobilized and released chemokines promote firm leukocyte adhesion and arrest by acting through G-protein–coupled receptors to activate leukocyte β2 integrins. Platelets can synthesize and release PAF, which can activate leukocyte αMβ2. CCL5 and the CXC chemokines ENA-78 and GRO-α, released by activated platelets, can also activate leukocytes. The chemokine neutrophil-activating peptide-2 (NAP-2) can be produced by the action of leukocyte cathepsin G on β-thromboglobulin secreted by platelets.762,763 Activated αMβ2 on leukocytes can interact with platelet GPIbα764 as well as with platelet-bound fibrinogen via a region(s) on the γ chain (amino acids 190 to 202765 and 377 to 395). Thrombospondin may serve as a bridging molecule between CD36 (GPIV) receptors, which are expressed on both platelets and mononuclear cells.766 Platelets also have intercellular adhesion molecule (ICAM)-2 on their surface, which is a ligand for the leukocyte integrin receptor αLβ2; although this ligand–receptor interaction appears to have only a minor role in platelet–leukocyte adhesion, it may be more important in leukocyte tethering.763 Platelet junctional adhesion molecule (JAM) 3 has also been suggested as a counterreceptor for leukocyte αMβ2.767 The immunoreceptor tyrosine-based activation motif (ITAM)-associated receptors GPVI and C-type lectin-like receptor-2 (CLEC-2) also promote platelet–leukocyte interactions during inflammation via their respective counterreceptors matrix metalloproteinase inducer (EMMPRIN) on neutrophils and macrophages and podoplanin on inflammatory macrophages.
Transcellular metabolism of eicosanoids can result in production of unique products (Fig. 2–10), and leukocytes can modify platelet activation.768 In a complementary fashion, the intimate relationship between leukocytes and platelets allows the latter to contribute to the inflammatory response, including the release of chemokines that can activate leukocytes; PDGF can affect fibroblast and smooth muscle cells; TGF-β1 both stimulates and inhibits cellular growth; and PF4 primes neutrophils and has antiangiogenic activity. Platelets synthesize the cytokine IL-1β, an important mediator of the inflammatory response.769 Platelets contain FcγIIA receptors that can localize IgG and immune complexes, resulting in complement activation. Platelets express CD40L on their surface after activation, and this molecule can interact with CD40, a member of the tumor necrosis factor (TNF) receptor family, on leukocytes and endothelial cells, leading to their activation and their elaboration of a number of proinflammatory molecules770–772 (see “CD40 Ligand [CD40L, CD154] and CD40”). Platelet CD40L also promotes procoagulant activity in endothelial cells.773 Finally, platelet–leukocyte interactions can promote the generation of reactive oxygen species, but platelets can also generate signals to stop their production.774
Figure 2–10.
Select aspects of transcellular eicosanoid metabolism. At sites of platelet–white blood cell (WBC) interactions, free arachidonic acid (AA) can be generated by both activated platelets and leukocytes and exchanged between the cells. In the platelet, cyclooxygenase 1 (COX-1), the target for aspirin, generates the major AA metabolite prostaglandin (PG) G2, the precursor for PGH2 that, in turn, is converted by thromboxane (TX) synthase to TXA2. TXA2 and PGH2 promote platelet activation and inflammation through binding to thromboprostanoid (TP) receptors. TXA2 is rapidly converted to TXB2. Platelets also express platelet-type 12-lipoxygenase (LOX), which converts AA to the relatively unstable intermediate 12-hydroperoxy-5,8,10,14-eicosatetraenoic acid (12-HPETE), which is subsequently converted to 12-hydroxyeicosatetraenoic acid (12-HETE). Platelets from most mammalian species do not possess 5-LOX and, therefore, cannot generate leukotriene A4 (LTA4) from AA. However, LTA4 produced by leukocytes can be transferred to interacting platelets, where it can be metabolized by glutathione-S-transferase to LTC4 or by platelet 12-LOX to the antiinflammatory mediator lipoxin (LXA4). In endothelial cells, AA can also be released from membrane phospholipids, but unlike in the platelet, it is sequentially metabolized by COX-1 or COX-2 and prostacyclin synthase to PGI2, which inhibits platelet activation by effects on the platelet inhibitory prostanoid (IP) receptor. Endothelial cells can also serve as a source of PGH2 that is metabolized by PGE synthase to PGE2. At high concentrations, PGE2 inhibits platelet activation, and at lower concentrations (<10–6 M), it activates platelets through the EP3 receptor. (Used with permission of Matt Hazzard, Teaching and Academic Support Center, The University of Kentucky.)
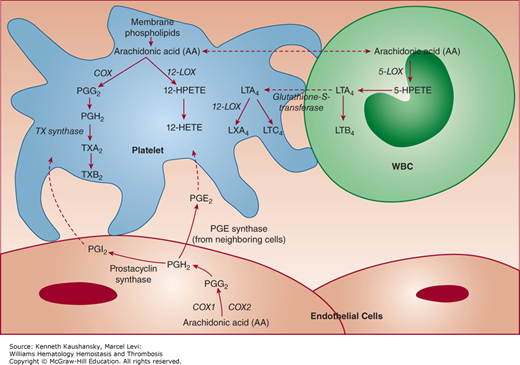
Platelet–leukocyte interactions may be important in the initiation of coagulation and fibrin formation through a P-selectin–dependent pathway. In fact, platelet–leukocyte aggregates facilitate thrombin generation to a greater extent than either platelets or leukocytes alone.775,776 Coincubation of platelets and leukocytes generates tissue factor activity, in part, through P-selectin–PSGL-1 interactions. The induction of tissue factor activity involves both de novo protein synthesis and exposure (“deencryption”) of latent tissue factor. The latter may occur by P-selectin–mediated production of tissue factor containing microparticles from leukocytes. Real-time imaging of platelet thrombus formation in vivo indicates that tissue factor accumulates in growing thrombi before leukocytes become associated with the thrombus. The accumulation of tissue factor and fibrin formation in thrombi depend on both platelet P-selectin and PSGL-1. These observations, coupled with the finding of bloodborne tissue factor antigen in the circulation,777 have led to a model in which platelet P-selectin recruits tissue factor-containing leukocyte microparticles to platelet-rich thrombi.778 Neutrophil-derived microparticles express active integrin αMβ2, which can interact with platelets by binding to GPIbα. This, in turn, can initiate platelet P-selectin expression, which will enhance the interactions with neutrophil microparticles containing the counterreceptor PSGL-1.779 In mice, increases in soluble P-selectin levels promote a procoagulant state associated with elevated levels of leukocyte-derived microparticles,780 and a P-selectin–immunoglobulin chimeric molecule can increase levels of leukocyte-derived microparticles in vitro and normalize the bleeding time in hemophilia A mice.781
Several clinical observations support a potential role for platelet–leukocyte interactions in vascular disease, including the presence of circulating platelet–leukocyte aggregates in patients with unstable angina782 and after coronary artery angioplasty783; in the latter situation, the presence of such aggregates appears to confer a worse prognosis for ischemic vascular complications.783 Circulating platelet–leukocyte aggregates are perhaps the most sensitive indicator of systemic platelet activation, reflecting the expression of P-selectin on the surface of platelets.784 Analysis of polymorphisms of PSGL-1 involving variable numbers of tandem repeats indicates that the longer PSGL-1 molecules are better able to form platelet–leukocyte aggregates; in some, but not all, studies, the longer molecules were associated with increased risk of some forms of thrombotic vascular disease.785–790 The S100 calcium-modulated protein family member MRP-14 (also known as S100A9), which is abundant in neutrophils and released by activated platelets, promotes platelet thrombus, at least in part through CD36.791
Platelets can contribute to both innate and adaptive immunity in several ways. Bacterial endotoxin binding to toll-like receptors can activate platelets (see “Toll-Like Receptors 1, 2, 4, 6, 9”), enhance platelet–neutrophil interactions, and promote bacterial trapping by stimulating the production of neutrophil extracellular traps (NETs) composed of DNA, histones, and enzymes that degrade pathogens.792–794 The production of NETs confers resistance to a variety of pathogens, including gram-positive (Staphylococcus aureus, Streptococcus pneumoniae, and group A streptococci) and gram-negative (Salmonella typhimurium, Shigella flexneri, and Escherichia coli) bacteria. A number of gram-positive bacteria can activate and aggregate platelets, and the platelet immune receptor RcγRIIA, integrin αIIbβ3, Src, and Syk, along with PF4, ADP, and TXA2, all play a role in the process.795 Platelets release mitochondria, which are related to bacteria in composition, when activated either in microparticles or free into plasma, where they associate with neutrophils and the platelet enzyme PLA2 IIA, which hydrolyzes mitochondrial and bacterial membranes, releasing a variety of proinflammatory molecules, including mitochondrial DNA, arachidonic acid, and lysophospholipids that are themselves capable of initiating NET formation.796 Release of platelet mitochondria during storage for transfusion has been suggested as being a contributor to platelet-associated nonhemolytic transfusion reactions.796
Thrombocytopenia is often present in association with bloodborne bacterial infections (sepsis), and the severity of the thrombocytopenia mirrors the severity of the infection and prognosis. Platelet factor V contributes to resistance to group A streptococcal infection797 by promoting thrombin generation and fibrin deposition, which may help to wall off the bacteria.797 Platelets also influence the function of lymphocytes.798 They enhance cytolytic T-cell proliferation and antibody production by B cells. Platelets can inhibit the responses of helper T cells and, via release of TGF-β1, increase regulatory T (Treg) cells. Finally, platelets can bind to malarial-infected erythrocytes and both suppress the growth of the parasites and destroy the intraerythrocytic malarial parasites.799
Platelets are essential to maintain the integrity of the vasculature, especially in inflammatory sites, although the mechanisms are not fully understood. Platelets store a number of barrier-stabilizing cytokines and growth factors that may be released constitutively or in a stimulus-dependent manner, including sphingosine-1-phosphate (S1P), which is essential for barrier function, ADP, serotonin, VEGF, and thrombospondin. While platelet G-protein–coupled signaling is essential for hemostasis and thrombosis after vascular injury, these pathways do not appear to be required for hemostasis during inflammation. And functional platelet ITAM motif receptors, CLEC-2 and GPVI, are required to maintain vascular integrity during inflammation, likely by triggering a unique response in the setting of inflammation.800
The partitioning between lymphatic and blood vessels during development requires normal platelet function. Platelets regulate lymphangiogenesis, at least in part, through interactions between platelet CLEC-2 and podoplanin on lymphatic endothelial cells. In addition, downstream ITAM signaling, mediated by Syk, SLP-76, and PLCγ2, is also required. Platelet activation along lymphatic endothelium may result in secretion of angiogenic factors. Importantly, platelet adhesion may result in intravascular hemostasis that promotes the lymphovenous junction, in that mouse embryos lacking CLEC-2, podoplanin, Syk, or SLP-76 display blood-filled lymphatic vessels. The requirement for platelets in maintaining blood–lymphatic separation extends beyond embryogenesis into adulthood. Importantly, the requirements for lymphovenous hemostasis are different from arterial and venous hemostasis, likely because of the low-flow, low-shear environment and intact lymphatic endothelium.
Platelet membrane glycoproteins mediate most of the interactions between platelets and their external environment. Receptors can receive signals from outside the platelet and transmit signals inside. In addition, glycoprotein receptors receive signals from inside the platelet that affect their external domain functions. Platelet glycoprotein receptors are grouped into several different receptor families (integrins, leucine-rich glycoproteins, immunoglobulin cell adhesion molecules, selectins, tetraspanins, and seven-transmembrane domain receptors; see Table 2–4). One member of the integrin family, integrin αIIbβ3, is virtually unique to platelets (and their precursors, megakaryocytes), whereas the leucine-rich glycoproteins GPIb/IX and GPV appear to have highly restricted but not uniquely platelet expression patterns, including cytokine-activated endothelial cells.801,802 All of the other receptors are expressed more widely on other cell types.
| Mr | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Gene Family | Common Name | Platelet Chain Designation | Integrin Designation | VLA† Designation | CD† Designation | Nonreduced | Reduced | Amino Acids | Carbohydrate | Lipid | Phosphorylated | Chromosome | Ligands | Platelet Specific | Function | Molecules on Platelet Surface (S) or Internal (I) | ||
| Integrin | Fibrinogen/receptor | αIIbβ3 | αIIbβ3-CD41a αIIb-CD41b β3-CD61 | αIIb β3 | 145,000 90,000 | αIIb α αIIb β | 125,000 23,000 114,000 | αIIb 1039 β3 762 | + + | – – | – + | 17 17 | Fib, VWF, Fn, Vn, ?TSP | + + | Adhesion, aggregation, protein trafficking | (S) 80,000 (I) 40,000 | ||
| Collagen receptor | GPla/IIa | α2β1 | VLA-2 | α2-CD49b β1-CD29 | α2 β1 | 150,000 138,000 | 148,000 | α2 1152 β1 778 | 5 10 | Collagen | – – | Adhesion | (S) 1000 | |||||
| Fibronectin receptor | CPIc*/IIa | α5β1 | VLA-5 | α5-CD49e β1-CD29 | α5 β1 | 140,000 138,000 | 148,000 | α5 1008 β1 778 | 12 10 | Fn | – | Adhesion | (S) 1000 | |||||
| Laminin receptor | GPlc/IIa | α6β1 | VLA-6 | α6-CD49f β1-CD29 | α6 β1 | 140,000 138,000 | 148,000 | α6 1067 β3 778 | 2 10 | Laminin | – | Adhesion | (S) 1000 | |||||
| Vitronectin receptor | αv/GPIIIa | αvβ3 | αv-CD51 β3-CD61 | αv β3 | 150,000 90,000 | αv αv | 125,000 25,000 114,000 | αv 1048 GPIIIa 762 | + | – | 2 17 | Vn, Fib, VWF, Fn, ?TSP, Osp | – | ?Adhesion, ?Protein trafficking | (S) 100 | |||
| Leucine-rich repeat glycoproteins | von Willebrand factor receptor | GPlb/Ix | Ib/Ix-CD42 Ib/α-CD42b Ib/β-CD42c Ix-CD42a | GPIb
GPIX | 170,000
17,000 | GPIbα GPIbβ | 145,000 22,000
17,000 | GPIbα 610(8)* GPIbβ 181(1)* GPIX 160(1)* | + + + | – + + | – + | 1 22 3 | VWF, thrombin | +? +? +? | Adhesion (high shear), ?thrombin activation | (S) 25,000 (S) 25,000 (S) 25,000 | ||
| GPV | GPV | 82,000 | 82,000 | GPV 544(15)* | + | + | + | 3 | +? | (S) 12,500 | ||||||||
| Immunoglobulin family cell adhesion modecules | PECAM-I | CD31 | 130,000 | PECAM-1 738 | + | ? | + | 17 | Heparin | – | ?Adhesion | (S) 8000 | ||||||
| Fcγ-RII | CD32 | 40,000 | FcγRII 324 | + | + | 1 | Immune complexes | – | Immune complex binding | (s) ~1000 | ||||||||
| HLA-Class 1 | HLA | + | 6 | – | Histocompatibility | (S) | ||||||||||||
| ICAM-2 | CD102 | 59,000 | ICAM-2 274 | 17 | LFA-1 | – | Platelet-leukocyte adhesion | (S) 2600 | ||||||||||
| GPVI | 62,000 | 65,000 | GPVI 316 | + | – | ? | Collagen | + | Activation | (S) ~2000 | ||||||||
| IAP | CD47 | 50,000 | IAP 287 | + | 3 | TSP | – | Activation | ||||||||||
| Selectins | P-Selectin (GMP 140; PADGEM) | CD62P | 140,000 | P-Selectin 830 | + | + | + | 1 | Sialyl-lex PSGL-1 | Platelet-leukocyte adhesion | (I) 20,000 | |||||||
| Tetraspanins | p24 | CD9 CD63 | 24,000 | CD9 228 | + | ? | – | Activation | (S) 40,000 | |||||||||
| PETA-3 | CD151 | 27,000 | CD151 253 | + | – | – | 11 | ? | – | Activation | (I) ~2000 | |||||||
| Lamp 3 (granulophysin) | CD63 | 53,000 | Lamp 3 238 | + | (I) 10,000 | |||||||||||||
| Miscellaneous | GPIV | CD36 | 88,000 | GPIV 471 | + | + | 7 | Collagen, TSP | – | Adhesion | (S) 20,000 | |||||||
| CLEC-2 | CD94 | CLEC-2 229 | + | + | 12 | Podoplanin | – | Adhesion/activation | ||||||||||
| TLR(1-6) | TLR | Pathogen-associated molecular patterns | – | Activation | ||||||||||||||
| Lamp 1 | CD107a | 110,000 | Lamp 1 389 | + | 13 | ? | – | ? | (I) 1200 | |||||||||
| Lamp 2 | CD107b | 120,000 | Lamp 2 381 | + | X | ? | ||||||||||||
| 67 kDa Laminin receptor | 67,000 | 67 kDa ?295 | X | Laminin | – | Adhesion | ||||||||||||
| ADP P2X1 receptor | 70,000 | P2X1 399 | + | 17 | ATP, ADP | – | Activation | (S) 13–130 | ||||||||||
| Leukosialin, sialophorin | CD43 | 90,000 | CD43 400 | + | + | 16 | ICAM-1 | – | Adhesion | |||||||||
| Seven-transmembrane domain (G protein-linked) | PAR-1 | 70,000 | PAR-1 425 | 5 | Thrombin | – | Activation | (S) ~1800 | ||||||||||
| PAR-4 | PAR-4 385 | + | + | 19 | Thrombin | – | Activation | |||||||||||
| Thromboxane A2 receptor | 55,000 | TXA2 343 | 19 | PGH2/thromboxane A2 | – | Activation | ~200 | |||||||||||
| α2-Adrenergic receptor | 64,000 | α2-Adrenergin 450 | 10 | Epinephrine | – | Activation | ~250 | |||||||||||
| Vasopressin receptor | 125,000 | Vasopressin 418 | ?x | Vasopressin | – | Activation | ~75 | |||||||||||
| ADP P2Y1 receptor | P2Y1 373 | + | 3 | ADP | – | Activation | ||||||||||||
| ADP P2Y12 receptor | P2Y12 342 | 3 | ADP | + | Activation | |||||||||||||
| Serotonin 5-HT2A | 53,000 | 5-HT2A | + | + | 13 | Serotonin | – | Activation | ||||||||||
Integrin receptors are heterodimeric complexes composed of an α subunit containing three or four divalent cation binding domains and a β subunit rich in disulfide bonds. Both subunits are transmembrane glycoproteins and are coded by different genes. There are at least 18 α subunits and eight β subunits.43,803,804 Three major families of integrin receptors are recognized based on the β subunit: β1, β2, and β3. Integrins are widely distributed on different cell types, and each integrin demonstrates unique ligand-binding properties. Integrin receptors mediate interactions between cells and proteins or proteins on cells; they are also involved in protein trafficking in cells. Integrin receptors can also transduce messages from outside the cell to inside the cell and from inside the cell to outside the cell.
The integrin αIIbβ3 complex, a member of the β3 integrin receptor family, is the dominant platelet receptor, with 80,000 to 100,000 receptors present on the surface of a resting platelet (Fig. 2–11).805–812 Another 20,000 to 40,000 receptors are present inside platelets, primarily in α-granule membranes, but also in dense bodies and membranes lining the open canalicular system; these receptors are able to join the plasma membrane when platelets are activated and undergo the release reaction.813–815 On average, integrin αIIbβ3 receptors are less than 20 nm apart on the platelet surface and thus are among the most densely expressed adhesion/aggregation receptors present on any cell type.
Figure 2–11.
αIIbβ3 Integrin structure and activation. A. Model for αIIbβ3 integrin inside-out activation and outside-in signaling. The α subunit is in blue, and the β subunit is in red. The bent, inactive receptor is depicted in (A). Under resting condition, the integrin β3 cytoplasmic domain appears to interact with filamin. Cellular stimulation induces migfilin to displace filamin from the integrin β3 cytoplasmic domain as well as a conformational change in talin that alters the interactions between the talin head and rod domains and exposes the talin head domain. The FERM F3 domain in the head then binds to the integrin β3 cytoplasmic domain, which unclasps the inter-subunit cytoplasmic and transmembrane domains from their complex with the integrin β3 cytoplasmic and transmembrane domains. Kindlin-3 binding to the integrin β3 cytoplasmic domain may facilitate talin binding and appears to be required for the conversion to the high-affinity state. The binding of talin then leads to separation of the ectodomain subunit tails and may diminish the interaction of the integrin headpiece with the tails. Although small ligands can bind to the receptor without headpiece extension, the large glycoprotein ligands may require extension to facilitate access to the ligand binding site. Extension (B) may occur spontaneously after leg separation or may result from traction force exerted on the integrin β3 cytoplasmic domain via talin’s association with the cytoskeleton and actin-myosin contractile force. Ligand binding to the integrin is associated with a swing out motion of the integrin β3 hybrid domain from the βA(I) domain (C), which results in both increased ligand affinity via alterations in the ADMIDAS (adjacent to metal ion-dependent adhesion site) and MIDAS (metal ion-dependent adhesion site) regions of integrin β3 and greater leg separation. This conformational change may initiate outside-in signaling. The ligated integrins may then cluster (not shown). The structure in panel (A) is based on the crystal structure of the ectodomain (PDB 3FCS)250 and the nuclear magnetic resonance (NMR) structure of the transmembrane and cytoplasmic domains (PDB 2K9J).894 The structure in (B) is based on the same ectodomain crystal structure, but with extension at the genus of the subunits (PDB 3FCS),250 the NMR structures of the separated transmembrane and cytoplasmic domains,894 and the structure of the complex between the β3 cytoplasmic domain and the talin F3 domain (PDB 2H7E).896 The structure in (C) is based on crystal structure of the liganded receptor (PDB 2VDN) headpiece,827 the extended structure of ectodomain (PDB3FCS),250 and the monomeric transmembrane structures connected to unstructured cytosolic tails. B. Domain structure of structure of integrin αIIbβ3. The individual domains and the ligand binding pocket are identified in the model of the extended integrin. I-EGF, integrin epidermal growth factor; PSI, plexins, semaphorins, integrins. C. The integrin transmembrane complex. Selected views of the NMR structure of the αIIb (red) and β3 (blue) transmembrane complex. The left panel depicts contacts involved in the outer membrane clasp, and the right panel depicts the contacts involved in the inner membrane clasp. Note that after the integrin αIIb helical region ends at V990, the next 5 residues (GFFKR) reenter the membrane; the two aromatic F residues make hydrophobic contacts with β3, and αIIb R995 makes a salt bridge with integrin β3 D723. (A, reproduced with permission from Lau TL, Kim C, Ginsberg MH, et al: The structure of the integrin alphaIIbbeta3 transmembrane complex explains integrin transmembrane signalling, EMBO J 2009 May 6;28(9):1351–1361. B, reproduced with permission from Zhu, J., et al: Structure of a complete integrin ectodomain in a physiologic resting state and activation and deactivation by applied forces, Mol Cell 2008 Dec 26;32(6):849–861. C, Reproduced with permission from Lau TL, Kim C, Ginsberg MH, et al: The structure of the integrin alphaIIbbeta3 transmembrane complex explains integrin transmembrane signalling, EMBO J 2009 May 6;28(9):1351–1361.)
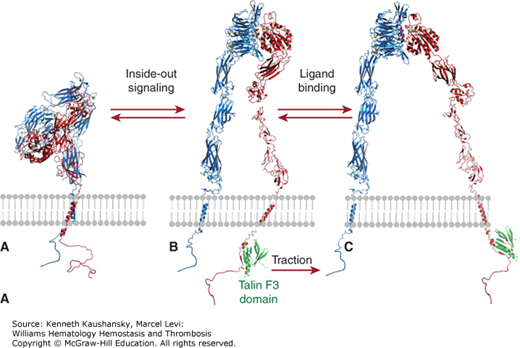
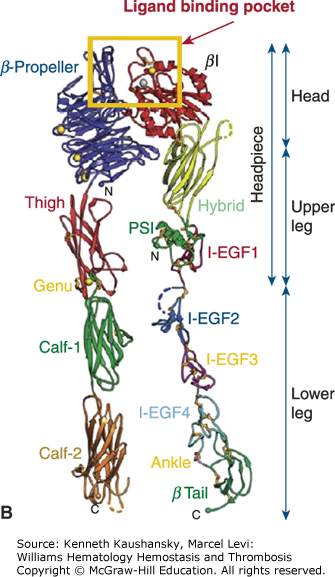
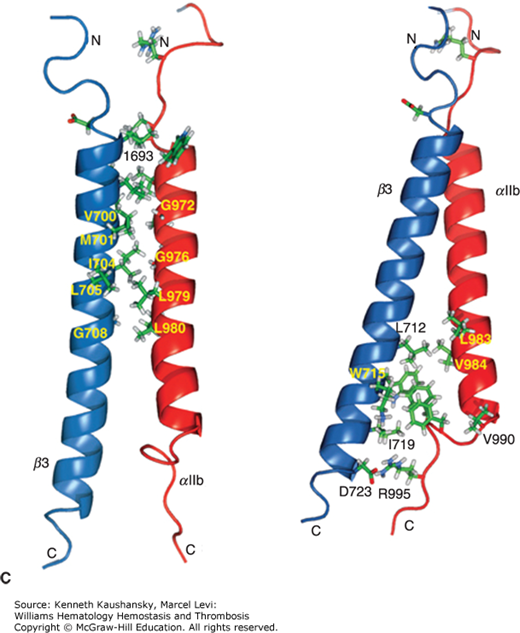
On resting platelets, integrin αIIbβ3 has low affinity for fibrinogen in solution, but when platelets are activated with ADP, epinephrine, thrombin, or other agonists, integrin αIIbβ3 binds fibrinogen relatively strongly.808,816 Activation induces changes in the integrin αIIbβ3 receptor itself that are responsible for the change in fibrinogen-binding affinity, but changes in the microenvironment surrounding integrin αIIbβ3 may also be involved. The integrin αIIbβ3 receptors in α granules appear to cycle to and from the plasma membrane.817 This recycling helps to explain the ability of the integrin to take up fibrinogen from plasma and transport it to α granules, where it is concentrated.375,818
Data from other integrin receptors identified a cell recognition sequence composed of RGD in the ligand fibronectin,819,820 and this same sequence is important in ligand binding to integrins αVβ3 and αIIbβ3. Fibrinogen contains one RGD sequence near the carboxy terminus of each of the two Aα chains (amino acids 572 to 574) and another at amino acids 95 to 97.821 In addition, the carboxyterminal 12 amino acid region of each of the two γ chains (amino acids 400 to 411) contains a sequence that includes Lys-Gln-Ala-Gly-Asp-Val, which is the most important in the binding of fibrinogen to platelets.822–826 VWF contains an RGD sequence in its carboxyterminal domain, and that region mediates the binding to integrin αIIbβ3.809,810,812 Small, synthetic peptides containing the RGD or γ-chain sequence inhibit the binding of fibrinogen to platelets, and these observations have been exploited to produce therapeutic agents (tirofiban and eptifibatide) to inhibit platelet thrombus formation827 (Chap. 24). Similarly, monoclonal antibodies that inhibit binding of ligands to integrin αIIbβ3 have been developed, and a mouse/human chimeric Fab fragment of one of them has been developed into a drug (abciximab) that is an effective antiplatelet agent.
The binding of fibrinogen to integrin αIIbβ3 appears to be a multistep process808,828–833: (1) the initial interaction is most likely via the γ-chain carboxyterminal region(s) and divalent cation-dependent823–826; (2) subsequent interactions enhance the binding and internalization of the fibrinogen834 and render it irreversible, even when divalent cations are removed835; (3) binding of fibrinogen induces changes in the receptor that can be recognized by antibodies (ligand-induced binding sites [LIBSs])442,826; (4) binding of fibrinogen to integrin αIIbβ3 induces changes in fibrinogen (receptor-induced binding sites) that can be recognized by antibodies and may involve exposure of the Aα chain Arg-Gly-Asp-Phe sequence at amino acids 95 to 98836,837; and (5) fibrinogen binding induces receptor clustering.251,838
By electron microscopy, the receptors have a globular head of 8 × 12 nm and two 18-nm long tails representing the carboxyterminal regions of each subunit, including their hydrophobic transmembrane domains.839,840 Crystallographic, electron microscopic, electron and neutron scattering, and biochemical data from integrin αIIbβ3 and the related integrin αVβ3 receptor indicate that the unactivated receptors are in a bent conformation and that activation involves both extension of the receptor head and a swing out motion in the β3 subunit.149,827,841–853 A three-dimensional reconstruction of integrin αIIbβ3 in a lipid bilayer nano disc from negative-stain electron microscopy images supports a compact conformation of the inactive receptor, but unlike the crystal structure of the ectodomain, the legs are not parallel and straight.848
Integrin αIIbβ3 shares the same basic structural features of all integrin receptors (see Table 2–4).30,848 The α subunit, αIIb, is a transmembrane protein with four characteristic divalent cation-binding sites (see Fig. 2–11). The mature protein contains 1008 amino acids43,854 with one transmembrane domain; during processing, it is cleaved into a heavy chain and a light chain connected by a disulfide bond. The β subunit, β3, contains 762 amino acids and is rich in cysteine residues, with a characteristic cysteine-rich region near its transmembrane domain.43,855 The integrin αIIb and β3 cytoplasmic tails consist of 20 and 47 amino acids, respectively. The genes coding for αIIb and β3 are very close to each other on chromosome 17 at q21.32, but are not so close as to share common regulatory domains.856,857 Both proteins are synthesized in megakaryocytes and join to form a calcium-dependent, noncovalent complex in the rough endoplasmic reticulum.858 Calnexin probably serves as a chaperone for integrin αIIb,859 but it is unclear which chaperone(s) are involved in integrin β3 folding and/or integrin αIIbβ3 complex formation. The integrin αIIbβ3 complex subsequently undergoes further processing in the Golgi apparatus, where the carbohydrate structures undergo maturation and the pro-GPIIb molecule is cleaved into its heavy and light chains by furin or a similar enzyme.860,861 Approximately 15 percent of the mass of both integrins αIIb and β3 are composed of carbohydrate.862 The mature integrin αIIbβ3 complex is then transported to the plasma membrane or the membranes of α granules or dense bodies. If integrins αIIb and β3 do not form a proper complex, either because of a structural abnormality in either subunit or the failure to synthesize one of the subunits, the subunit(s) that are synthesized are rapidly degraded and so are not expressed on the membrane surface (Chap. 11). Degradation of integrin αIIb appears to involve retro-translocation from the endoplasmic reticulum into the cytoplasm, ubiquitination, and proteolysis by the megakaryocyte proteasome.859
Both integrins αIIb and β3 are composed of a series of domains (see Fig. 2–11). The aminoterminal region of integrin αIIb contains a seven-blade β-propeller domain, and each blade is composed of four β strands connected by loops. The propeller interacts with the βA (I-like) domain of integrin β3, forming the globular head region observed in electron micrographs. The four calcium ions bound by the propeller domain interact with β hairpin loops in blades four to seven that extend away from the interface with integrin β3. In addition, there is a unique integrin αIIb cap subdomain made up of four loops from blades one to three that are unique to αIIb and contribute to its ligand binding specificity. The remainder of the extracellular components of integrin αIIb are made up of a thigh, genu (knee-like), and two calf domains,250 much like the structure of the related integrin αV subunit.841,844 The cytoplasmic domain of integrin αIIb interacts with the cytoplasmic domain of integrin β3 and the interaction is important in controlling activation of the holoreceptor.863–866 The cytoplasmic domain of integrin αIIb has a GFFKR sequence near the membrane that is thought to control inside-out activation of the integrin receptors because mutations or deletions in this region result in the receptor adopting a conformation with high affinity for fibrinogen.867–871 A number of studies using mutagenesis and nuclear magnetic resonance (NMR) identified different structures for the transmembrane and cytoplasmic domains, and differences in the relative roles of heterodimeric and homodimeric associations.864,872–875 Disrupting the conformation of this region also results in a constitutively high-affinity receptor,876,877 which has led to the conclusion that inside-out activation of integrin αIIbβ3 requires separation of the transmembrane and cytoplasmic domains, but it remains possible that more subtle changes in the cytoplasmic and transmembrane domains may be sufficient.848
The integrin β3 subunit domains are not linearly arranged because the first domain (PSI [plexins, semaphorins, and integrins]) was subjected to the insertion of a hybrid domain, which itself was subjected to the insertion of a βA (I-like) domain; the latter domain is homologous to the VWF A domain and integrin I domains, both of which bind ligands (see Fig. 2–11).827,878 The double insertion in the PSI domain explains why there is a “long range” disulfide bond extending from C13 to C435; thus, even though the βA domain makes contact with the integrin αIIb propeller (via Arg261 and other residues that interact with two rings of hydrophobic residues in the integrin αIIb “cage”), it is not the aminoterminus of the molecule. The PSI domain contains Leu33, which defines the PlA1 (HPA-1a) specificity, as opposed to the alloantigen PlA2 (HPA-1b), which is produced by a Pro33 polymorphism. The integrin β3 leg is composed of four integrin EGF domains that are rich in disulfide bonds. In the crystal structure, this region interacts with the integrin αIIb stalk region and the globular head in the bent, unactivated receptor, but these interactions are less prominent in the three-dimensional reconstruction of the inactive receptor not in the activated receptor.250,827,848 Mutations in the integrin EGF domains, including cysteine residues, can activate the receptor as can the binding of monoclonal antibodies.879–882 The importance of the normal disulfide bond pairings in integrin β3 is further supported by data demonstrating that certain reducing agents can cause activation of integrin αIIbβ3, fibrinogen binding, and platelet aggregation,883,884 and an enzyme capable of catalyzing the exchange of thiol groups and disulfide in proteins (PDI) has been identified on the surface of platelets and in platelet releasates.883,885–887 Thiol-disulfide exchange in integrins αIIbβ3 and αVβ3 is implicated as a contributor to clot retraction.888 Moreover, regions in integrin β3 itself have the same consensus sequence (CGXC) present in PDI that is thought to mediate the catalysis.889 One model suggests that integrin αIIbβ3 can achieve a low level of activation without alterations in disulfide bonds, but that maximal activation requires PDI or similar activity along with a source of thiols such as plasma glutathione or a membrane NAD(P)H oxidoreductase system.883 Inhibition of PDI and other enzymes that mediate thiol-disulfide exchange (ERp57, ERp5) reduces platelet thrombus formation.890,891 It is still unclear, however, whether disulfide bond alterations contribute to activation in vivo under physiologic or pathologic conditions.
Transmembrane domain structures of integrin αIIb and integrin β3 have been proposed based on NMR and structural modeling studies.871,873,874,892–896 Because the integrin αIIb transmembrane helix is shorter than the integrin β3 helix, they traverse the membrane at an angle of approximately 25 degrees. The association of the integrin αIIb and integrin β3 ectodomains near the site of entry into the membrane results in the transmembrane helices being directly juxtaposed in the region of the membrane closest to the ectodomain (outer membrane clasp). Near the cytoplasmic end of the membrane, the helices are held together by an inner membrane clasp composed of the integrin αIIb residues immediately after the end of the helix (GFFKR), with the membrane reimmersion of F992 and F993 filling the gap and interacting with integrin β3 W715 and I719, with integrin αIIb R995 creating a salt bridge with integrin β3 723 and perhaps residue 726.897,898 Of note, these regions are conserved in many other integrins receptors, and so the basic mechanism may be common to many of the receptors.
Inside-out signaling is accomplished by the talin F3 domain binding to the integrin β3 cytoplasmic domain, which is proposed to disrupt the inner membrane clasp.34,244,245,863,865,866,869,870,872,876,892,899,900 This may be facilitated by migfilin displacing filamin from the integrin β3 cytoplasmic domain as the latter interaction may prevent talin binding.901 Talin binding results in dissociation of the transmembrane helices and reorganization of the cytoplasmic region of integrin β3 into a more extended helix. Integrin αIIbβ3 ectodomain chain separation, headpiece extension, and integrin β3 swing out then follow, either spontaneously or as a result of the traction force generated by the cytoskeleton on integrin β3 through talin.149 Outside-in signaling is presumed to be initiated by loss of ectodomain interactions between the membrane-proximal regions of integrins αIIb and β3, perhaps as a result of ligand binding producing even greater integrin β3 swing out, resulting in disruption of the outer membrane clasp and subsequent dissociation of the transmembrane helices. This potentially may facilitate the interaction of the cytoplasmic domains with cytoskeletal elements and signaling molecules.
The integrin β3 tail also contains two NXXY motifs, and Y747 and Y759 within one of these motifs are phosphorylated upon platelet aggregation, thus producing docking sites for signaling molecules.235 Studies in mice and in recombinant systems demonstrate a role for the sites in clot retraction and platelet aggregate stability.291,902
A number of proteins have been shown to bind to the cytoplasmic domains of integrin αIIb and/or β3, either directly or through interactions with other proteins, including signaling molecules (Src, Shc, FAK, paxillin, and ILK, all of which bind to integrin β3), cytoskeletal proteins (kindlin-3, skelemin, α-actin, and myosin, which bind to integrin β3, and filamin and talin, which bind to integrins αIIb and/or β3), and other proteins (β3-endonexin and CD98, which bind to integrin β3, and CIB and calreticulin, which bind to αIIb) (Fig. 2–12).244,866,903–919 These interactions are important in mediating inside-out signaling and outside-in signaling.235 JAM-A is a negative regulator of outside-in activation by integrin αIIbβ3 that acts by regulating activation of Src.920 Similarly, PECAM-1 serves as an inhibitor of integrin αIIbβ3 activation through a sequential phosphorylation mechanism.921,922 Force on the integrin β3 cytoplasmic domain by actin–myosin action may supply the energy for the conformational change in integrin αIIbβ3 from bent to extended.250
Figure 2–12.
Protein interactions with the cytoplasmic domains of αIIbβ3 regulate inside-out and outside-in signaling. Shown are some, but not all, of the proteins reported to associate with the αIIbβ3 cytoplasmic domains, many in a dynamic fashion. Some are associated with resting platelets, while others are recruited to, or dissociate from, the integrin during inside-out or outside-in signaling, leading to F-actin assembly. In addition, several proteins with enzymatic function become activated (asterisks) after fibrinogen binding to αIIbβ3. Not shown are the many additional adapter molecules, enzymes, and substrates that may become recruited through more indirect interactions. CIB, calcium and integrin-binding 1; Csk, c-Src tyrosine kinase; ILK, integrin-linked kinase; ITAM, a yet-to-be identified protein with one or more immunoreceptor tyrosine activation motifs; PKCβ, protein kinase Cβ; PP1c, protein phosphatase 1c; RACK1, receptor for activated C kinase 1; Syk, spleen tyrosine kinase. (Reproduced with permission from Coller, B.S. and S.J. Shattil, The GPIIb/IIIa (integrin alphaIIbbeta3) odyssey: a technology-driven saga of a receptor with twists, turns, and even a bend, Blood 2008 Oct 15;112(8):3011–3025.)
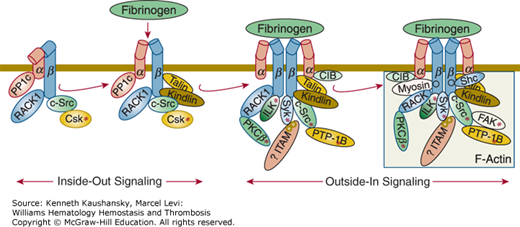
The junction between the integrin αIIb propeller and the β3 βA (I-like) domain is the site of ligand binding to integrin αIIbβ3 (see Fig. 2–11). This region of integrin β3 contains three divalent cation binding sites: MIDAS (metal ion-dependent adhesion site), ADMIDAS (adjacent to MIDAS), and SyMBS (synergy metal binding site).250 The latter was previously termed the ligand-associated metal binding site (LIMBS) based on the crystal structure of integrin αVβ3.844,845
The crystal structure of integrin αVβ3 demonstrated that an RGD peptide is bound primarily via interactions between the Arg in the peptide and two Asp residues (D150 and D218) in integrin αV and between the Asp in the peptide and the MIDAS cation.845 The binding pocket in integrin αIIbβ3 is similar but differs in that only one Asp in integrin αIIb (D224) is available to interact with an Arg (or Lys as in the fibrinogen γ-chain peptide), the distance between D224 in integrin αIIb and the MIDAS cation is longer, and a cap subdomain of the integrin αIIb propeller contributes Phe160 to a hydrophobic exosite in combination with Tyr190.149,827 As a result, the pocket is able to accommodate the longer fibrinogen γ-chain C-terminal peptide better, with the peptide’s Asp and C-terminal Val carboxyls interacting with the MIDAS and ADMIDAS cations, respectively.826 It also explains why integrin αIIbβ3 can bind peptides containing the longer Lys residue (KGD peptides).923 Crystal structures are also available for the integrin αIIbβ3 receptor with the drugs eptifibatide and tirofiban, which are effective antithrombotic agents because of their ability to block ligand binding to integrin αIIbβ3, and demonstrate specificity for integrin αIIbβ3 compared to integrin αVβ3.827 The basis of the specificity of these agents involves in part their interaction with the integrin αIIb-specific exosite and the greater length between their positive and negative charges.827 The third integrin αIIbβ3 antagonist drug, abciximab, is a chimeric murine monoclonal antibody Fab fragment. Its epitope has been localized to a region on integrin β3 very close to the MIDAS, suggesting that it works by steric interference with ligand binding, disruption of the binding pocket, or both mechanisms.
Two major conformational changes in integrin αIIbβ3 have been described in association with activation: headpiece extension and integrin β3 hybrid and PSI domain swing-out (see Fig. 2–11).250,827,853 Headpiece extension can contribute to ligand binding by enhancing access to the binding site; it can also contribute to platelet aggregation by extending the receptor out further from the platelet surface,924 thus facilitating the ability of fibrinogen to bridge between platelets.846 The integrin β3 hybrid and PSI domain swing-out motion appears to enhance ligand binding, but the precise mechanism is unclear.826,847,850 Swing-out is associated with movement of the ADMIDAS metal ion and the α1–β1 loop toward the MIDAS with the latter movement stabilized by the interaction of two backbone nitrogens in the α1–β1 loop with the ligand carboxyl oxygen, thus reinforcing the binding to the MIDAS metal ion.149,826 Mutations that produce swing-out of the hybrid and PSI domains result in constitutive ligand binding to integrin αIIbβ3.925
Binding of fibrinogen to platelet integrin αIIbβ3 leads to platelet aggregation, presumably via crosslinking of integrin molecules on two different platelets by fibrinogen.840 The dimeric and relatively rigid structure of fibrinogen and the location of the binding sites at the ends of the γ chains are all consistent with such a model as the two binding sites on a single fibrinogen molecule are probably more than 45 nm apart. Soon after fibrinogen binds, it can be dissociated from the platelet by chelating the divalent cations, but the binding becomes irreversible within an hour.835 Fibrinogen binding alone is not sufficient for platelet aggregation, but the events necessary after fibrinogen binding, which probably include ligand- and/or cytoskeletal-mediated receptor clustering, are not well understood.95,835,926,927 After ligands bind to integrin αIIbβ3, “outside-in” signaling through the integrin can occur, resulting in a number of phosphorylation events, changes in the platelet cytoskeleton, platelet spreading, and even initiation of protein translation.236,237,928
In addition to fibrinogen, several other proteins can bind to integrin αIIbβ3 on activated platelets, including VWF, fibronectin, vitronectin, thrombospondin, and prothrombin390,675,929; each of these contains an RGD sequence in the region implicated in the initial interaction with platelets. There are subtle differences in the binding of each of these ligands, however, with regard to divalent cation preference and competent activating agents. The binding of all of these other ligands can also be inhibited by RGD-containing peptides, indicating a common requirement for the interaction between the RGD sequence in the protein and the RGD-binding site in integrin αIIbβ3.930,931
Platelet aggregation measured in the aggregometer ex vivo depends upon fibrinogen binding to integrin αIIbβ3. It is less clear whether fibrinogen is the most important ligand supporting platelet aggregation in vivo since studies performed in model systems under flowing conditions indicate that VWF is the major ligand at higher shear rates.932 Even in the aggregometer, VWF can partially substitute for fibrinogen if the fibrinogen concentration is very low.933 In vivo, mice deficient in both VWF and fibrinogen still make platelet thrombi in response to vascular injury.934–936 Although fibronectin was initially implicated in supporting the development of such thrombi, mice deficient in fibrinogen, VWF, and fibronectin have paradoxically increased platelet aggregation and thrombus formation, suggesting that fibronectin may play an inhibiting role in thrombus formation under certain circumstances.373
Although resting platelets do not bind soluble fibrinogen (or other adhesive glycoproteins) to an appreciable extent, they can adhere to fibrinogen immobilized on a surface.825,937 This activation-independent adhesion may be from alterations in the structure of fibrinogen when it is immobilized on a surface.836,938 Alternatively, there may always be a few integrin αIIbβ3 receptors that are transiently in the proper conformation to bind fibrinogen, and immobilization may result in high local density of fibrinogen and favorable kinetics for adhesion. Finally, it is possible that even low-affinity fibrinogen interactions with integrin αIIbβ3 are sufficient to initiate integrin interactions with the cytoskeleton such that actin-myosin–induced contraction provides the energy required for the conformational changes needed to achieve higher affinity binding.250
Fibrinogen and/or fibrin have been identified on the surface of damaged blood vessels; thus, it is possible that integrin αIIbβ3 mediates platelet adhesion under those circumstances.939 In contrast, integrin αIIbβ3 on resting platelets does not appear to be able to mediate adhesion to VWF or fibronectin940; if platelets are activated, however, integrin αIIbβ3 can support adhesion to these glycoproteins.930 In models of platelet accumulation under flowing conditions, αIIbβ3 acts in synergy with GPIb/IX, VWF, and fibrinogen at the apex of thrombi, where shear forces are greatest.28,941,942 The integrin αIIbβ3 has also been implicated in platelet spreading after adhesion,227,228,943 and it is necessary for clot retraction (see above) and the uptake of plasma fibrinogen into platelet α granules.818,944
Less well-defined roles for integrin αIIbβ3 have been suggested in the binding of plasminogen,688 calcium transport across the platelet membrane,945–947 IgE binding to platelets leading to parasite cytotoxicity,948 and interactions with the Borrelia species spirochetes that cause Lyme disease949 and hantavirus.950 Integrin αIIbβ3 also mediates factor XIIIa binding to platelets, but this is primarily as a result of factor XIII’s association with fibrinogen.456 Factor XIIIa and calpain have also been implicated in limiting platelet–platelet interactions after activation by adhesion to collagen.951
Integrin α2β1 (GPIa/IIa) is widely distributed on different cell types and can mediate adhesion to collagen.19,20,952–957 The integrin α2 subunit (GPIa) contains a region of 220 amino acids inserted in the aminoterminal β-propeller region (I domain) that is homologous to similar regions in other proteins that are known to interact with collagen, including VWF and cartilage matrix protein.958 This region has a MIDAS, and crystallographic data of the α2 I domain in complex with a CRP containing the type I collagen sequence GFOGER (where O indicates hydroxyproline) demonstrated that the glutamic acid in the peptide coordinates Mg2+ binding in the MIDAS.959–961 The integrin α2β1 I domain can assume a variety of conformations, going from inactive (closed), through intermediate or low affinity, to active high affinity.952,962
Both integrin α2β1 and GPVI appear to participate in platelet interactions with collagen.963–965 Bleeding defects have been described in patients with decreased levels of integrin α2β1 and GPVI, but the precise contributions of the decreases in these receptors is uncertain (Chap. 11). Although integrin α2β1 is capable of supporting adhesion to collagen without exogenous activators, like integrin αIIbβ3, it appears to be able to increase its affinity for ligand in response to inside-out activation.966,967 Potential initiators of integrin α2β1 activation include signaling after GPVI interaction with collagen and GPIb-mediated adhesion to VWF, perhaps acting via actin polymerization.959,968–970 Thus, one possible scenario is that following GPIb-mediated adhesion to VWF and collagen adhesion and activation mediated by GPVI, integrin α2β1 may promote firm adhesion to collagen, stabilize thrombus growth on collagen, and promote procoagulant activity.971,972 In addition, the affinity of integrin α2β1 may also be modulated by alterations in disulfide bonds since inhibition of platelet PDI and sulfhydryl blocking agents inhibit integrin α2β1-mediated platelet adhesion to type I collagen and to the related peptide GFOGER.883,973 The state of the collagen may also influence whether integrin α2β1 or GPVI mediates the interaction with collagen, because GPVI appears to mediate adhesion to fibrillar collagen, whereas integrin α2β1 preferentially adheres to collagen that has been treated with partial protease digestion.28,974
Ligand binding to integrin α2β1 is enhanced in the presence of magnesium or manganese and is inhibited by calcium, and thus the conditions in human blood, where calcium concentrations are higher than those of magnesium, do not provide optimal cation concentrations for the receptor’s function.975 Integrin α2β1 can, however, mediate platelet adhesion to collagen in heparinized blood,956,975 and inhibitors of integrin α2β1 inhibit thrombus formation in animal models.976–978 Regions of collagen type I have been implicated as potential binding sites for integrin α2β1979; the peptide sequence 502 to 516 of collagen type I α1 chain, which contains a Gly-Glu-Arg (GER) sequence, may be of particular importance,980 but other regions of the collagen molecule may also be important.981 In type III collagen, amino acids 522 to 528 of fragment α1 (III) CB4 contain a binding region for α2β1.982
Three different alleles for the integrin α2 gene, which differ at nucleotides 807 (T or C) and 1648 (G or A), have been described.983 The 807 substitution does not affect the amino acid sequence, but the 1648 substitution causes a change from Glu to Lys, resulting in the Brb and Bra alloantigens (HPA-5a and HPA-5b). Allele 1 (T-G) is present in 39 percent of individuals, allele 2 (C-G) in 53 percent, and allele 3 (C-A) in 7 percent.984,985 Individuals with allele 1 have higher integrin α2β1 platelet density than individuals with allele 2, and individuals with allele 3 have the lowest density; the density of integrin α2β1correlates with platelet deposition on collagen under flow. The association of these polymorphisms with cardiovascular disease morbidity and mortality, including the risk of developing MI986,987 and stroke,988 has been extensively study without firm conclusions, although there is some suggestion that they may be associated with cardiovascular risk.983,989–992
Integrin α2β1 is probably linked to the membrane skeleton.993 Its ligand specificity appears to be determined by the cell on which it is expressed, since on endothelial cells it functions as a laminin receptor as well as a collagen receptor.994,995 Engagement of integrin α2β1 is capable of initiating platelet protein synthesis.236 Integrin α2β1 has been implicated in megakaryocyte development and platelet formation. In particular, loss of activated integrin α2β1 receptors on the surface of megakaryocytes, as a result of interacting with collagen, has been implicated in the transition from the marrow to the peripheral circulation,967 and conditional targeting of megakaryocyte and platelet integrin α2β1 in mice is associated with reduced MPV.996
Integrin α5β1 is a receptor that is expressed on a wide variety of different cells and mediates adhesion to fibronectin.804,819,820 It is important for interactions with extracellular matrix, and data from cells other than platelets indicate a role for this receptor in developmental biology and metastasis formation. The RGD sequence in fibronectin is crucial for cell adhesion, but other regions in fibronectin probably also contribute. RGD-containing peptides can inhibit cell adhesion mediated by integrin α5β1. As with other integrin receptors, adhesion depends on the presence of divalent cations. Integrin α5β1 is competent to mediate adhesion of resting platelets to fibronectin,997,998 but its affinity may be modulated by activation.999 The biologic role of this receptor on platelets is not clear. Although it may be involved in hemostasis and/or thrombosis, it is also possible that its function is primarily related to megakaryocyte binding to marrow matrix and proplatelet formation.1000 Integrin α5β1 is not the only fibronectin receptor on platelets, since with appropriate activation, integrin αIIbβ3 can also bind fibronectin.804,1001
Platelet adhesion to select laminins, which are variably found in basement membranes and extracellular matrix, can be mediated by integrin α6β1.804,1002–1004 Because VWF can bind to some laminins, GPIb can also contribute to platelet adhesion to laminin.1002 This adhesion is best demonstrated with magnesium and manganese; calcium does not support adhesion. This receptor is competent on resting platelets, but its role in platelet physiology is not clear. Mice deficient in integrin α6β1 do not bleed pathologically but are protected against thrombosis.1002 The integrin appears to be able to signal in platelets via PI3 kinase to induce morphologic changes.1005 An approximate Mr 67,000 laminin receptor has also been identified on platelets; this receptor is present on other cells as well.1006
Integrin αVβ3 receptor shares the same β subunit as integrin αIIbβ3 (GPIIb/IIIa) (see Fig. 2–11).804,855,1007–1009 The integrin αV and αIIb subunits display 36 percent sequence identity.1010 Integrin αVβ3 differs dramatically, however, from integrin αIIbβ3 in its platelet surface density, because there are only approximately 50 to 100 integrin αVβ3 receptors per platelet.1011 The crystal structure of the external domains of integrin αVβ3 alone and in complex with a peptide containing the RGD cell recognition sequence found in a number of ligands has been solved at high resolution.844,845 Such RGD peptides inhibit ligand binding to integrin αVβ3. The most important findings were: (1) the receptor adopts a bent conformation in which the globular headpiece composed of the N-terminal β-propeller region of αV and the βA (I-like) domain of integrin β3 lies near the legs of the integrin αV and β3 subunits, and (2) the RGD peptide binds to the headpiece with the Arg (R) making contact with integrin αV and the Asp (D) making contact with the MIDAS domain in β3. Current evidence suggests that the bent conformation is the inactive one and that activation results in extension of the headpiece and pivoting between the integrin β3 βA and hybrid domains in association with leg separation.827,843,1007,1009 Integrin αVβ3 can mediate adhesion to vitronectin, but only in the presence of magnesium or manganese, not calcium.1011 It can also mediate interactions with fibrinogen, VWF, prothrombin, and thrombospondin.389,1012–1015 Platelet stimulation can activate integrin αVβ3, analogous to activation of integrins αIIbβ3 and α2β1. Activated integrin αVβ3 may uniquely mediate adhesion to osteopontin, a protein found in high concentrations in atherosclerotic plaque.1016 The receptor’s role in platelet physiology is not defined, but it may contribute to the development of platelet coagulant activity.1017
The integrin αVβ3 receptor is also present on endothelial cells,822,1013 osteoclasts,1018 smooth muscle cells, and other cells; it has been implicated in bone resorption,1019–1021 endothelial–matrix interactions,822,1013 lymphoid cell apoptosis,1022 neovascularization,1023 tumor angiogenesis,1023–1025 intimal hyperplasia after vascular injury,1026–1028 sickle cell disease,1029–1031 focal segmental glomerulosclerosis,1032,1033 and scleroderma.1034
The presence or absence of integrin αVβ3 on the platelets of patients with Glanzmann thrombasthenia can help localize the abnormality to either integrin αIIb (if integrin αVβ3 is present in normal or increased amounts) or integrin β3 (if integrin αVβ3 is reduced or absent) (Chap. 11).
GPIb is composed of GPIbα (CD42b) (610 amino acids) disulfide-bonded to two GPIbβ subunits (CD42c) (122 amino acids).801,1035–1043 GPIb appears to exist on the surface of platelets in a 1:1 complex with GPIX (160 amino acids) and a 2:1 complex with GPV (Fig. 2–13). The GPIbα gene is on the short arm of chromosome 17, and the GPIbβ gene is on the long arm of chromosome 22. The GPIX gene is on the long arm of chromosome 3.1044–1046 GPIX is required for efficient surface expression of GPIb,1047 but beyond that, its function is unknown. GPIb/IX is expressed on megakaryocytes and platelets; there is controversy as to whether GPIb/IX is expressed on endothelial cells, either constitutively or after cytokine activation.802 The promoters for GPIb/IX lack TATA or CAAT boxes, but contain binding sites for the GATA and ETS families of transcription factors, which, along with the expression of the cofactor FOG (friend of GATA-1), may account for the limited expression of GPIb/IX.1048–1056
Figure 2–13.
The organization of GPIb/IX complex. GPIbα (green), GPIbβ (blue), and GPIX (purple) subunits are colored differently. Left: A cartoon illustration of the GPIb/IX complex largely drawn in ribbon diagrams. Various parts of GPIbα are labeled on the left. Right: The top view of the membrane-proximal portion of GPIb/IX that contains the stalk region of GPIbα, the extracellular domains of GPIbβ and GPIX, and a portion of the transmembrane (TM) helical bundle. The disulfide bonds between GPIbα and GPIbβ are highlighted in red. Side chains of Tyr106 in GPIbβ are shown in blue spheres, one of which is located at the interface 1 between GPIbβ and GPIX. Residue Pro74 in GPIbβ is shown in orange spheres, one of which is located at or close to the interface 2. (Reproduced with permission from Li R, Emsley J: The organizing principle of the platelet glycoprotein Ib-IX-V complex. J Thromb Haemost 2013 Apr; 11(4):605–614.)
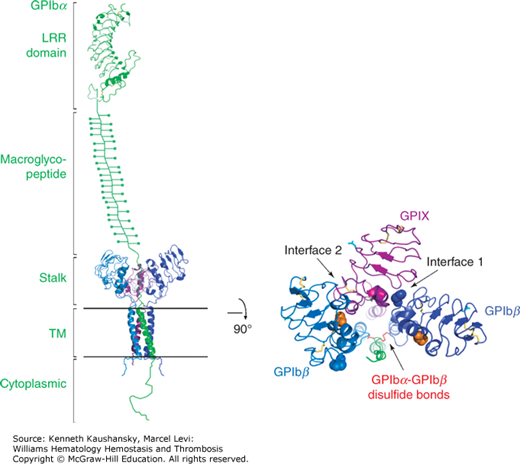
A genetic polymorphism in GPIbα affects the number of repeating 13-amino-acid units (1, 2, 3, or 4) and produces changes in the molecular weight of GPIbα.1057 The 2 repeat variant is most common, but there is considerable ethnic variation in the frequency of the different numbers of repeats. This molecular weight polymorphism has been linked to the Sib and Ko alloantigens, which have been localized to a T→M variation at amino acid 145 of GPIbα, with M associated with either 3 or 4 repeats and T associated with either 1 or 2 repeats.984 Some, but not all, reports suggest an association between the alleles with the larger number of repeats and vascular disease.983,991,1058,1059 Two other GPIbα polymorphisms have been described: (1) C or T at position –5 from the ATG start codon (RS system), and (2) a nucleotide dimorphism at the third bases of the codon for Arg 358.1038,1060,1061 A C at position –5 is present in only 8 to 17 percent of individuals and more closely resembles the sequence surrounding the ATG start codon (Kozak sequence) considered optimal for translation. In fact, this polymorphism is associated with higher levels of platelet surface GPIb and may be a risk factor for ischemic vascular disease.1062–1070 GPIb has been implicated as a target antigen in autoimmune thrombocytopenia and in quinine and quinidine-induced thrombocytopenia (Chap. 7).
GPIbα has a large number of O-linked carbohydrate chains terminating in sialic acid residues,1071 and the latter contribute significantly to the negative charge of the platelet membrane.215 Electron micrographic analysis indicates that GPIb exists as a long flexible rod (approximately 60 nm) with two globular domains of approximately 9 and 16 nm.1072 Thus, GPIb probably extends much further out from the platelet’s surface than does integrin αIIbβ3, which may account for its primacy in platelet adhesion, as well as the increased risk of cardiovascular disease in individuals with longer GPIb molecules because of an increased number of 13-amino-acid repeats. The long extension may also make it susceptible to conformational changes induced by shear forces.801 The extracellular region of GPIbα is readily cleaved by a variety of proteases, including platelet calpains,1073 yielding a soluble fragment named glycocalicin that circulates in normal plasma at 1 to 3 mg/L.1074 In vivo, platelet shedding of glycocalicin from GPIbα is mediated by a disintegrin and metalloprotease (ADAM)-17 (also termed TACE) cleaving a juxtamembrane sequence1075,1076; shedding is controlled by GPIbβ interactions with an unidentified protein, calpain, and reactive oxygen species.1077–1079 Levels of plasma glycocalicin correlate with platelet production and thus can be used to differentiate thrombocytopenia based on decreased platelet production from thrombocytopenia as a result of increased platelet destruction.1080–1085
GPIbβ and GPIX have free sulfhydryl groups in their cytoplasmic domains that undergo palmitoylation, at least in part, further anchoring the protein to the membrane.1086,1087 The penultimate serine residue at the C-terminus of GPIbα is phosphorylated, providing an attachment site for the signal-complex protein 14–3–3ζ.1088 Similarly, GPIbβ can undergo phosphorylation of Ser 166 in its cytoplasmic domain as a result of protein kinase A activation via cAMP, providing another binding site for 14–3–3ζ (see Fig. 2–13).1089–1091 The cytoplasmic domain of GPIbα connects GPIb to filamin A (actin-binding protein), thus connecting GPIb to the platelet cytoskeleton.993,1092,1093 Coordinated expression of GPIbα and filamin is required for efficient expression of both proteins, and imbalances result in abnormalities in platelet size.1094,1095 Alterations in the cytoskeleton can affect GPIb functional activity.1096–1098 14–3–3ζ can bind PI3 kinase and has been implicated in GPIb-mediated intracellular signaling that results in integrin αIIbβ3 activation; Lyn, Vav, Rac1, Alet, and Lim kinase-1 also have been implicated in GPIb/IX–mediated signaling.9,1099–1101 GPIb also appears to be in close proximity to FcγRIIA and the Fc receptor γ-chain, two receptors that can initiate signaling via tyrosine phosphorylation of their cytoplasmic ITAM sequences by Src family kinases and recruitment of the tyrosine kinase syk.1102–1105 Engagement of GPIb by VWF may lead to clustering of GPIb-IX–V complexes in glycolipid-enriched microdomains or lipid rafts, which may serve to concentrate signaling molecules; clustering also increases ligand avidity.1106
GPIbα has eight leucine-rich repeats in the aminoterminal region of its extracellular domain, whereas GPIbβ and GPIX have one each.1039,1042,1045 These repeats are consensus sequences of 24 amino acids with seven regularly spaced leucines; well-defined disulfide loop sequences flank the repeats.801 Similar leucine-rich repeats are present in a variety of other proteins.
Crystal structures of the N-terminus of GPIbα (amino acid residue 1–305) alone and in complex with native and mutated A1 domains of VWF provide important information on the interactions between these proteins (Fig. 2–14).1107,1108 This region of GPIbα adopts a curved shape made up of an N-terminal β-hairpin flanking sequence (finger) containing a C4-C17 disulfide loop (H1-D18), eight leucine-rich repeats (K19-W204), a β-switch region (V227-S241), and a C-terminal sulfated anionic region (D269-D287), with Y276, Y278, and Y279 undergoing posttranslation sulfation.1108–1110 The VWF-A1 domain, which has alternating β strands and α helices organized into a central β-sheet surrounded by amphiphatic α helices, interacts with the concave face of GPIbα with two areas of tight interactions, at the N-terminal β-hairpin + first leucine-rich repeat (with VWF A1 domain loops α1β2, β3α2, and α3β4), and a more extensive interaction at leucine-rich repeats 5 to 8 + the β switch region (with VWF A1 domain helix α3, loop α3β4, and strand β3). The structure of the VWF A1 domain when not bound to GPIbα differs from that of the bound VWF A1 in that the α1β2 loop protrudes in a way that would prevent interaction with GPIbα.1108 This observation and others related to differences in the ability of different-sized fragments of VWF and GPIbα to interact indicate that other regions of both proteins probably contribute to both the binding and activation of the receptor. The crystal structure of GPIbα with the naturally occurring mutation M239V in the β-hairpin region that results in platelet-type (pseudo-) von Willebrand disease (Chap. 16) has also been obtained,1109 and demonstrates a more stable β-hairpin conformation, which probably accounts for the approximately sixfold increase in binding affinity, primarily through an increase in the association rate. Leucine-rich repeats 3 to 5 do not demonstrate interaction with the normal VWF A domain in the crystal structure, but they are important in ristocetin-induced platelet agglutination and platelet adhesion at high shear; they do participate to some extent in crystal structures with gain-of-function mutations in VWF A1.1107,1111,1112 It has been proposed that hydrodynamic forces produced at high shear alter the A1 domain and expose regions that interact with these repeats in GPIb.1113 Other natural and site-directed mutations causing the platelet-type von Willebrand disease pattern of enhanced VWF binding (G233V, V234G, D235V, K237V) also affect the β-hairpin region. A number of Bernard-Soulier syndrome mutations that cause loss of VWF binding to GPIbα localize to the concave face of leucine-rich repeats 5, 6, and 7 (L129P, A156V, and L179del) and to the sides of leucine-rich repeat 2 (C65R and L57P).1110
Figure 2–14.
Structural, binding, and mutational features of the A1 domain (cyan) bound to GPIbα (magenta). Disulfides are in yellow stick. The A1-GPIbα complex forms a super β-sheet at the interface between the A1 β3 and GPIbα β14 strands. Platelet-type von Willebrand disease (VWD) mutations (green Cα atom spheres) stabilize the β-switch in its bound over its unbound conformation. VWD type 2B mutations (red Cα spheres) locate distal from the GPIbα interface, near to the A2 termini where elongational force is applied. VWD type 2B mutations are hypothesized to stabilize an alternative, high-affinity conformation. A region of GPIbα that is important for interaction with A1 in high shear (leucine-rich repeats [LRR] 3–5) and with ristocetin is shown in gray. (Adapted with permission from Li R, Emsley J: The organizing principle of the platelet glycoprotein Ib-IX-V complex. J Thromb Haemost 2013 Apr; 11(4):605–614.)
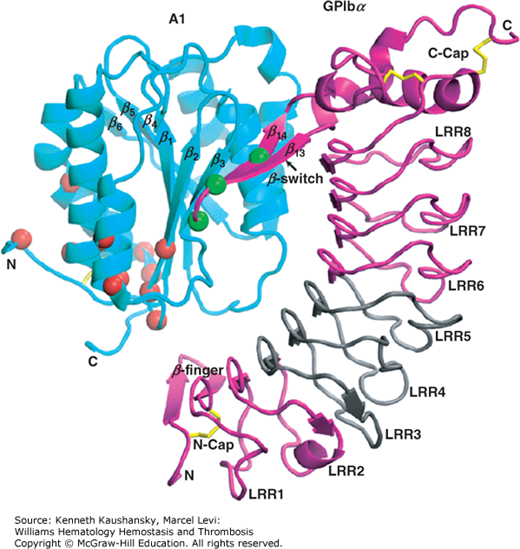
The GPIb ectodomain crystal structure has been determined, confirming the four predicted conserved disulfide bonds (C1-C7, C5-C14, C68-C93, and C70-C116), along with the unpaired C122, which crosslinks to GPIbα.1114 The two former disulfides are in the N-capping region, and the two latter are in the C-capping region flanking the single leucine-rich repeat.1040 Using a chimeric GPIbβ/GPIX ectodomain protein, the likely contacts between GPIb and GPIX were identified. The structure proposed is a tetramer of one GPIbα, two GPIbβs, and one GPIX in which GPIX interacts with one of the GPIbβ molecules.1037,1040,1043
Plasma VWF will not bind to GPIb under static conditions unless the antibiotic ristocetin or the snake venom botrocetin is added. The mechanism by which ristocetin induces VWF binding to GPIb is unclear, but effects on VWF as well as on platelet surface charge have been described, and dimerization of ristocetin molecules and multimerization of VWF, as well as stabilization of an A1 domain conformation with high affinity for GPIb, have also been implicated.801,1113,1115–1118 Botrocetin binds to VWF, exposing the site that binds to GPIb.1119 Peptide studies implicate the anionic, sulfated tyrosine region of GPIb as the binding site for botrocetin-treated VWF.801
Unlike integrin αIIbβ3, which requires intact, activated platelets to bind to VWF, GPIb-mediated VWF binding does not require platelet activation or even platelet metabolic integrity, because fixed platelets are readily agglutinated in the presence of VWF and either ristocetin or botrocetin.1116 This observation forms the basis of the assay of plasma VWF activity.
Platelets will adhere to VWF when the latter is immobilized on a surface, even in the absence of ristocetin or botrocetin.1116,1120–1122 Under these circumstances, the VWF is believed to undergo a conformational change that allows for direct interactions. It may not, however, be necessary to propose a change in VWF conformation as the interaction between VWF and GPIb appears to have both high association and dissociation rates, permitting tethering and translocation on a surface coated with a high density of VWF, but minimal interaction in fluid phase.809 Similarly, VWF associated with fibrin can interact with platelet GPIb without ristocetin or botrocetin.61,1123 The C1C2 domains of VWF appear to contain a fibrin binding site.304
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


