Fig. 8.1
pDC plasticity. During viral infection and certain autoimmune diseases (Lupus, Psoriasis) pDCs contribute to immune response through TLR-7/9 engagement by viral or endogenous nucleic acid leading to their production of IFN-I and phenotypic maturation. However, in other contexts, such as airway or oral antigen delivery, in absence of TLR-7/9 engagement, pDC can also contribute to peripheral tolerance by favoring Treg differentiation and expansion
pDCs are essential for recognition of altered self and thus may contribute to immune responses directed towards transformed cells. It would, therefore, be expected that an increased presence of pDCs in tumor tissues should promote activation, immune recognition of tumor antigens and, in turn, lead to tumor rejection.
8.1.2 Evidence for a Role of Type-I IFN in Cancer Immunosurveillance
Although type-II IFN has emerged early as a key mediator of tumor immunosurveillance, it is only very recently that the role of IFN-I has been discovered [8–10]. Indeed, gene-targeted mice lacking the type-I IFN receptor (IFNAR1) develop more carcinogen-induced primary tumors than WT control mice [9, 11] and antibody-mediated blockade of the IFNα/β receptor in WT hosts abrogated rejection of immunogenic transplanted tumors [9]. Several recent reports have shown that host-derived IFN-I responses are critical in early steps of tumor immunosurveillance [8–10, 12] as well as in the efficacy of radiotherapy [13] or of certain chemotherapeutic treatments [14].
Moreover, in humans, exogenous administration of recombinant IFN-α has shown efficacy in the treatment of cancer patients [15] and high intratumor expression of IFN-I responsive genes has a positive prognostic value in cancers [16–18]. In line with this observation, it has been shown recently that downregulation of IFNAR1 in tumor stroma stimulates tumor development and growth, playing a key role in the formation of an immune-privileged niche, and predicting poor prognosis in human colorectal cancer patients [19].
In a mechanistic view, recent studies in mouse models have shown that IFNs contribute to anti-tumor immunity via stimulating specific CD8α+ DCs to cross-present tumor-derived antigens to cytotoxic T lymphocytes (CTLs) [8, 9]. In addition, IFN-I provides a signal to stimulate the clonal expansion of CTLs [20] and increase their viability [21]. Furthermore, recent studies have identified the cGAS/STING pathway of DNA sensing as a critical route for type-I IFN induction during cell transformation and chemo or radiotherapy treatment (for review [22]). STING agonists are currently in clinical trial aiming at increasing IFN-I production in the tumor to limit neoplastic growth and promote specific anti-tumoral immune responses. However, the nature of the IFN-I-secreting cells in most cancer models remains undetermined and other tumor-derived ligands and innate sensors are likely to contribute to induction of endogenous IFN-I. In this perspective, pDCs represent prime suspects with regard to their presence in the diseased tissue, their ability to sense host-derived factors through TLR7/9, and their IFN-I producing specialization.
8.1.3 pDCs in Breast Tumors
Breast cancers are the most common malignant tumors and the first leading cause of cancer death in women. Among several types of breast cancers, the so-called triple negative (non-amplified Her2neu, ERneg and PRneg) (TN) is the most aggressive one. Breast cancers are considered as immunogenic tumors as (1) CD8+ T cell responses as well as humoral responses against TAA (Her2neu, p53, Muc1) have been demonstrated [23–27] and (2) CD8+ T cell infiltration has been recently reported to correlate with better prognosis [28–30]. Moreover we recently demonstrated that primary breast tumors (BT) are largely infiltrated by immune cells involved in innate sensing, i.e., NK cells, dendritic cells (DC), and macrophages (MΦ) that display an activated phenotype [31, 32] suggesting their stimulation within the BT environment. We and others also reported the presence of strong CD4+ [33] and CD8+ [24–27] T lymphocytes infiltrates in BT suggesting that all the players required to set up an efficient anti-tumor response are present within the BT environment. However, when tumors are clinically detected, this immune response is, in most cases, unable to counteract cancer development because tumors have developed immunosubversion processes.
Several studies have pinpointed that the tumor microenvironment subverts the function of immune cells and favors immunosuppression, avoiding the establishment of anti-tumor immunity. Recent works including from our group [32, 34] have also shown that pDC accumulate in several types of solid cancers, but very limited studies analyzed their function within the tumor microenvironment.
In this context, we have observed that BT infiltration by pDC is associated with an adverse clinical outcome [32], suggesting that they might contribute to the tumor immune evasion and ultimately to its outgrowth.
In a prospective study analyzing 79 newly diagnosed breast cancer patients, we observed a preferential accumulation of pDC in aggressive BT with a high mitotic index and a TN phenotype [35]. These results strengthen our previous observation on the deleterious impact of TApDC on breast cancer patients’ outcome [32]. Such tumor-associated (TA)-pDC exhibited a partially activated phenotype (CD40, CD83, CD86, and HLA-DR intermediate levels) when compared to patients’ associated blood pDC. This partially activated phenotype may be related to the local action of GM-CSF on TApDC [36] or to endogenous nucleic acid as TLR7/9 ligands. Of note, the concomitant increase of GM-CSF and pDC was significantly associated with relatively more aggressive breast cancer subtypes. In addition, similarly to tonsil pDC, TApDC activated in vitro by TLR7 and 9 agonists retain their capacity to mature and induce the proliferation of naïve CD4+ T cells and their secretion of IL-10 and IFN-γ. In contrast, TApDC were strongly impaired in their ability to secrete IFN-α upon TLR7/9 stimulation in vitro. This functional defect was specific to IFN-α and occurred selectively at the tumor site. Indeed, the production of [1] inflammatory molecules such as CXCL10 in TApDC [35] as well as [2] IFN-α in patients’ blood pDC was not affected [35, 37].
8.1.4 pDCs in Ovarian Tumors
Ovarian cancer (OC) is the most frequent and aggressive gynecologic cancer. This is due, at least in part, to its diagnosis at advanced stages (III/IV) in the majority of patients with peritoneal carcinosis and malignant ascites [38, 39]. We investigated the clinical significance of the presence of pDC in tumor mass and malignant ascites by conducting a systematic comparison of pDC number, phenotype, and function in blood, tumor, and ascites. We observed an accumulation of pDC in most of malignant ascites and their presence at high frequency in 36% of primary tumors. Importantly, as in breast tumors, accumulation of pDC in tumors was an independent prognostic factor associated with early relapse.
These results obtained on 33 OC patients in whom pDC were identified by flow cytometry as CD4+BDCA2+CD123+ cells [38] were confirmed on a larger series of OC patients (n = 97) by immunohistochemistry (IHC) [39]. In this study, TApDC were identified as BDCA2+ cells on formalin-fixed, paraffin-embedded (FFPE) tissues using tissue microarray (TMA) allowing the analysis of the impact of the presence of pDC in both cancer epithelium and lymphoid aggregates present in cancer stroma. BDCA2+ TApDC were present in only 18/97 tumors (18%). In univariate analysis, the presence of TApDC within cancer epithelium was associated with early relapse and shorter median overall survival. In multivariate analysis, in addition to clinical prognostic factors (advanced stage, debulking surgery, and residual tumor), the presence of TApDC remains an independent prognostic factor associated with shorter PFS [39].
Thus, using two different and complementary methods (flow cytometry and IHC) on two independent cohorts, we observed a deleterious impact of the presence of TApDC within OC tumors on patient’s outcome. These data corroborate our findings in breast cancer [32] and others in melanoma [34] showing that TApDC accumulation correlates with poor prognosis. Collectively, these results suggest that TApDC may contribute to immune tolerance and tumor progression.
Like in breast tumors, we showed that, unlike ascites pDC, TApDC from ovarian cancer (1) expressed a semi-mature phenotype as evidenced by high levels of CD40 and CD86 and (2) were strongly impaired for their IFN-α production in response to CpG-A (TLR9 ligand) known to induce huge amounts of type-I IFNs [38]. These results suggest that TApDC respond to endogenous signals delivered by the tumor microenvironment by maturing rather than by producing IFN-α, suggesting a preferential activation of NFκB rather than IRF7 pathway [40, 41].
8.1.5 Mechanisms Leading to TApDC Functional Defect
The similarity in the biology of TApDC between breast [32, 35, 42] [5] and ovarian [38, 39] tumors strengthens the importance of our work and indicates that common inhibitory mechanisms could occur between epithelial cancers with different localizations.
In both breast and ovarian cancers, we demonstrate that besides IFN-α, the production of IFN-β and TNF-α but not CXCL10 nor MIP1-α/CCL3 by TLR-activated healthy pDC is impaired by the breast and ovarian tumor environments [38, 42]. Importantly, we identified TGF-β and TNF-α as major soluble factors involved in TApDC functional alteration (Fig. 8.2). Indeed, recombinant TGF-β1 and TNF-α synergistically blocked IFN-α production of TLR-activated pDC, and neutralization of TGF-β and TNF-α in tumor-derived supernatants restored pDCs’ IFN-α production [38, 42]. The involvement of tumor-derived TGF-β was further confirmed in situ by the detection of phosphorylated Smad2 in the nuclei of TApDC in breast tumor tissues [42]. Mechanisms of type-I IFN inhibition did not involve TLR downregulation but the inhibition of IRF-7 expression and nuclear translocation in pDC after their exposure to tumor-derived supernatants or recombinant TGF-β1 and TNF-α.
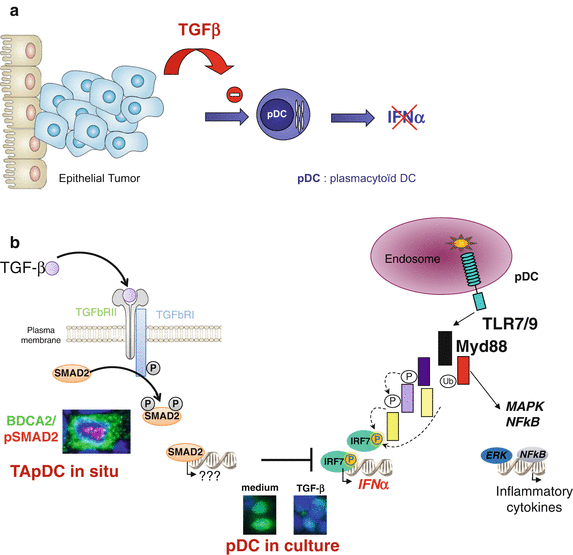
Fig. 8.2
TApDC inhibition of type-I IFN production through TGF-β. (a) TGF-β produced in breast and ovarian tumor environment potently suppressed type-I IFN production by pDC. (b) In situ TApDC expressed nuclear phosphorylated-Smad2 demonstrating their exposition to TGF-β, and leading to blockade of IRF7 nuclear translocation
Recently, E. Gatti and collaborators (CIML) showed that the molecule BAD-LAMP (LAMP5) is uniquely expressed by non-activated human pDCs among hematopoietic cells [43] and that BAD-LAMP controls TLR9 trafficking to LAMP1+ late endosomes, reducing IFN-I production in pDCs activated by CpG DNA and favoring TNF-α. Conversely, BAD-LAMP silencing allows spontaneous TLR9 activation in absence of exogenous TLR agonists. Together, we further demonstrated that breast TApDCs display increased BAD-LAMP expression that contributes to their inability to produce IFN-I. BAD-LAMP expression therefore limits IFN-I expression in pDCs by promoting TLR9 sorting to late endosomes at steady state and in response to TME (Combes, under revision). “BAD-LAMP controls TLR9 trafficking and signaling in human plasmacytoïd dendritic cells” by Alexis Combes, Voahirana Camosseto, Prudence N’Guessan, Rafael Argüello, Julie Mussard, Christophe Caux, Nathalie Bendriss-Vermare, Philippe Pierre, and Evelina GattiNat Com, in press
As IFN-I plays important anti-tumor functions, either directly or through immunosurveillance activation [44], our work suggests that the inhibition of TApDCs’ IFN-I production by BT might confer a selective advantage to tumor cells and represent a novel escape mechanism.
The alteration of IFN-α production by pDC in response to TLR ligands has been previously described in chronic viral infections such as HIV and HCV contributing to the failure of an efficient immune response [45]. In the context of cancer, previous works have also reported this alteration in breast [35], lung [46], and head and neck [47] cancers, and chronic myeloid leukemia [48].
Our findings indicate that targeting TApDC to restore their IFN-α production, by combining TLR7/9-based immunotherapy with TGF-β antagonist, might be an achievable strategy to induce anti-tumor immunity in breast cancer.
8.2 Treg in Breast Tumors
The presence of regulatory T cells (Treg) has been described in a large panel of solid tumors. However, their impact on tumor progression differs according to the tumor type analyzed [49]. We recently obtained evidence in breast carcinoma that Treg localized within lymphoid aggregates, but not in the tumor bed, have a negative impact on patients’ survival [33]. The poor impact of Tregs has been confirmed by others in invasive breast carcinoma [50] and the reduction of Treg during neoadjuvant chemotherapy correlates with improved prognosis [51]. Moreover, we showed selective Treg recruitment through CCR4/CCL22 in the lymphoid aggregates upon contact with mature DC, where they became strongly and selectively activated (HLA-DRhigh, inducible co-stimulatory molecule (ICOS)high) and block conventional T-cell response.
8.2.1 Treg Recruitment
Several studies including from our group demonstrated the infiltration of BT by immune subsets involved in immune tolerance, i.e., pDC [32] and CD4+CD25highCD127negFoxP3+ Treg [30, 33, 52] and type-2 MΦ [for review, see [53], Ramos RN et al. submitted] that are all of poor prognosis for overall survival (OS) in primary BT.
An in-depth ex vivo analysis demonstrated that tumor-associated Treg (TATreg) (1) are activated as they express ICOS, HLA-DR, GITR, and CTLA-4, (2) are functional as they suppress CD4+ T cells proliferation and IFNγ secretion, (3) proliferate in situ in contrast to the resting non regulatory CD4+ memory T cells and CD8+ T cells detected within BT [33].
In contrast to associated patients’ blood Treg, TATreg present a selective loss of membrane CCR4, consecutive to an active recruitment through CCL22 secreted within the BT environment [33]. In line with this, (1) CCL22, but not CCL17 induced the CCR4 downregulation and (2) BT lacking CCL22 expression are not infiltrated by TATreg independently of their production of CCL17, the other CCR4 ligand.
In primary BT context, independently of the molecular subtype of the tumor, CCL22 expression is strongly increased compared to peri-tumoral breast tissue as assessed not only by IHC but also by ELISA within the BT dilacerations supernatants [31].
Interestingly, at the systemic level, we observed a gradual increase in CCL22 plasmatic levels from healthy subjects, patients with primary BT, first metastatic relapse or with more advanced BT [54] that could reflect the tumor burden.
Using BT epithelial cell lines but also primary BT specimens, we demonstrated the major role of immune infiltrate in the selective induction of CCL22 but not CCL17 by tumor epithelial cells [31]. In vitro experiments using (1) inhibitory antibodies against cytokine receptors and/or cytokines or (2) exogenous recombinant cytokines demonstrate the preponderant role of a dialogue between epithelial tumor cells and tumor-infiltrating NK cells and MΦ for this CCL22 production [31]. Through these studies we propose the following sequence of events (Fig. 8.3): (1) NK cells detecting tumor cells secrete IFNγ, (2) IFNγ activates MΦ favoring their secretion of IL-1β and TNFα, (3) these 3 cytokines act together to increase CCL22 production by epithelial tumor cells. This was further confirmed in ex vivo experiments using primary BT specimens demonstrating the cooperation of MΦ and NK cells to favor CCL22 production by freshly purified tumor cells [31].
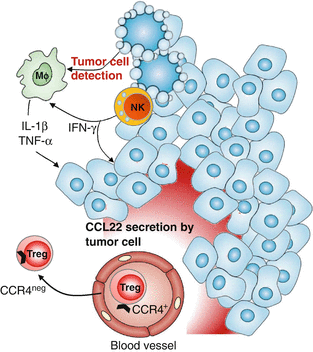
Fig. 8.3
IFNγ, TNFα, and IL-1β secretion by NK and MΦ leads to CCL22 secretion by transformed cells attracting CCR4+ Treg in breast tumor. Healthy breast epithelial cells secrete low levels of CCL22 in a polarized manner within the luminal acini, their transformation favor their recognition by infiltrating NK cells leading to IFNγ secretion. IFNγ promoted MΦ activation that will secrete TNFα and IL-1β after interaction with breast epithelial tumor cells. Combined action of IFNγ, IL-1β, and TNFα will induce strong CCL22 non polarized secretion by tumor cells that will induce the recruitment of CCR4+ Treg from periphery, leading to CCR4 internalization
This illustrates a mechanism allowing the transformed breast epithelial cells to counteract the local inflammation involving NK and MΦ to favor Treg recruitment through CCL22 secretion as previously described in chronically inflamed colon [55]. In turn, TATreg may also favor tumor progression via (1) the inhibition of NK cytolytic functions (for review [56]) (2) the conversion, as recently demonstrated in HIV context [57] of type-1 MΦ into type-2 MΦ that have pro-tumor functions through production of factors promoting angiogenesis, tumor cell proliferation, and favoring immunosuppression (for review [53, 58]).
All together, these data strongly indicate that CCL22 participates to the immunosubversion in BT and will favor disease progression. In this context, CCR4 antagonists (small molecule (AF-399)) have been validated to block in vitro and in vivo CCL22-mediated recruitment of human Treg and Th2 cells [59].
8.2.2 pDC-Mediated Treg Expansion
High numbers of TATreg is an independent factor of poor prognosis for BT patient’s survival. Furthermore, BT infiltrating CD4+FoxP3neg T cells (TATconv) are of memory phenotype (CD45RO+) and very few are activated contrasting with highly activated TATreg (ICOShigh, CTLA-4+, GITR+, HLA-DR+), that proliferate in situ (Ki67+) and display suppressive activity in vitro [33]. Furthermore, ICOS, belonging to the CD28 family is highly expressed by TATreg as reported in melanoma [60] and ovarian tumors [61]. Despite their Ki67 expression in situ, TATreg did not proliferate in vitro under classical stimulation with anti-CD3/anti-CD28 agonist Ab coated beads, in the presence of exogenous IL-2, in contrast to blood Treg or Tconv either from BT or blood that proliferated strongly. Of importance, infiltration of BT by pDC, also of poor prognosis for patients’ survival [32], correlates with Treg infiltration and both cell subsets co-localized within tumor [62] and especially within TNBT [35]. We thus wondered whether TApDC might contribute to TATreg expansion within the BT microenvironment. We observed that TApDC as well as healthy donors pDC preconditioned with BT-derived supernatants were very potent in inducing [1] the selective expansion of Foxp3+ Treg and [2] the differentiation of IL-10-secreting CD4+ T cells [62]. Interestingly, exogenous IFN-α reverted immunosuppressive CD4+ T cell responses induced by TApDC and BT environment [35], indicating that such TApDC tumor-promoting capacity is strongly amplified in tumors as a result of their impaired IFN-α production.
In order to understand the negative impact of TApDC, we developed an orthotopic murine mammary tumor model that closely mimics the human pathology, including pDC and Treg infiltration [63]. We showed that TApDC are mostly immature and maintain their ability to internalize antigens in vivo and to activate CD4+ T cells in vitro. Most importantly, TApDC are specifically altered for cytokine production in response to TLR9 ligands in vitro while preserving unaltered response to TLR7 ligands. In vivo pDC depletion delayed tumor growth and reduced intratumoral Treg frequency, showing that TApDC provide an immunosubversive environment most likely through Treg activation favoring tumor progression.
pDC were previously shown to regulate growth of multiple myeloma (MM) cells [64] and more recently to favor bone metastasis of breast cancer cells [65].
Whereas healthy donors pDC overexpressed ICOS-L, the unique ligand of ICOS, after in vitro activation by TLR7 or TLR9 agonists, TApDC despite their activated phenotype (CD40+HLA-DR+CD86+), lacked ICOS-L expression in breast and ovarian tumor dilacerates [38, 62]. Interestingly, in vitro ICOS-L engagement by activated ICOS+ CD4+ T cells led to its downregulation at pDC membrane and 24 h culture of tumor cell dilacerate suspensions in the presence of a blocking anti-ICOS mAb restored ICOS-L expression on TApDC [62]. These data demonstrate that ICOS/ICOS-L interaction occurs in BT during Treg/pDC contacts.
Interestingly, allogeneic reactions of [pDC + TA-CD4+ T cells] co-cultures led to a strong enrichment and proliferation of FoxP3+ Treg and enhanced IL-10 secretion. This immunosuppressive T cell response to pDC stimulation was highly dependent on ICOS as the addition of a neutralizing anti-ICOS mAb selectively inhibited both Treg proliferation and IL-10 secretion. Furthermore, myeloid DC (mDC, LinnegHLA-DR+CD11c+BDCA2neg) that did not overexpress ICOS-L after activation were not associated with Treg enrichment nor strong IL-10 secretion [62]. In agreement with our observations on TATreg, proliferation in response to pDC of ICOS+ Treg issued from ovarian tumor ascites [61] or ICOS expressing natural Treg (nTreg) from thymus [66] was also dependent on ICOS/ICOS-L interaction. In addition, pDC were also reported to increase IL-10 production by CD4 T cell through ICOS/ICOSL interaction [67, 68]. Furthermore, we recently confirmed, in collaboration with D Olive team, the in vivo downregulation of ICOS-L on Follicular Lymphoma B cells and the ICOS/ICOS-L-dependent expansion of Treg in this tumor environment [69].
Importantly, neither IL-2, IL-17 nor IFN-γ was detectable in BT dilacerates in contrast to IL-10 [62]. ICOS neutralization only slightly reduced IFN-γ secretion and proliferation of Tconv and did not impact T cell response to mDC. This suggests that ICOS favors TATreg expansion and IL-10 production but does not participate in the induction of immune effectors in primary BT. Of most importance, on a retrospective cohort of BT patients, ICOS expression was mainly detected on Treg by IHC and ICOS+ cell infiltration correlated with reduced PFS and OS in univariate analysis [62], in agreement with results in ovarian tumors [61].
ICOS constitutes a critical regulator of humoral immune responses, mainly as it stimulates follicular helper T cell (Tfh) activation, as illustrated in ICOS-deficient mice and patients [70]. This said, a few reports also suggest that ICOS may contribute to anti-tumor cellular immunity. Indeed, in melanoma patients, an increased proportion of IFN-γ-producing CD4+ICOS+ T cells has been observed in patients responding to anti-CTLA-4 (ipilimumab) treatment [71], and ICOS-deficient mice bearing B16 tumors do not respond properly to anti-CTLA-4 therapy.
ICOS+ Treg have been described in several human and mouse tumors and our in vitro experiments demonstrated that ICOS+ TATreg are strongly dependent on ICOS for their amplification, contrary to Treg from peripheral blood [62]. ICOS dependency could reflect a particular subpopulation of Treg, either linked to a particular origin similarly to thymic ICOS+ nTreg [66] or to their microenvironment and/or activation status. In this context, it is not clear whether TATreg are nTreg or are induced from naïve T cells in the periphery. Deciphering whether ICOS blockade displays a differential impact between nTreg, iTreg, and TATreg could be of importance in the perspective to revert TACD4+ T cell immunosuppressive response in breast cancer patients.
Taken together, our data suggest that ICOS blockade might be a promising strategy to eradicate TATreg and IL-10-producing CD4+ T cells in primary BT. The ICOS neutralization might need to be transient in order to abrogate Treg amplification while leaving unperturbed the restoration of effector cells potentially expressing ICOS [71].
8.2.2.1 Conclusion
Collectively our results show that BT microenvironment inhibits type-I IFN production by TApDC through TGF-β and TNF-α that confers them enhanced capacity to promote FoxP3high TATreg expansion and IL-10-secreting T cells via ICOS-ICOSL interaction in vivo. This favors the accumulation of immunosuppressive CD4+ T cells at the tumor site preventing anti-tumor immune responses (Fig. 8.4). Our observations pave the way for the development of new therapeutic strategies for breast cancer patients by (1) restoring TApDCs’ IFN-α production using a combination of TLR-7/9 ligands with TGF-β and TNF-α antagonists, for the induction of a potent anti-viral-like anti-tumor immunity, or (2) neutralizing ICOS/ICOS-L interaction between pDC and Treg.
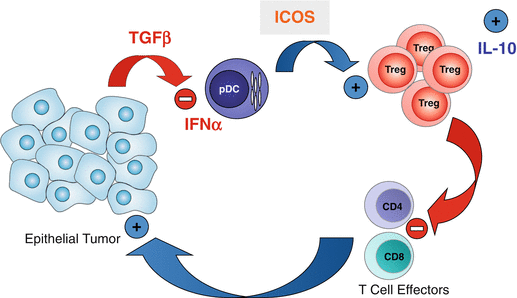
Fig. 8.4
pDC/Treg at the center of immunosuppressive networks in breast tumor environment, role of ICOS/ICOSL. Through ICOS-L expressed on TApDC, ICOS engagement participates in Treg expansion and IL-10 secretion in BT and ICOS+ cells are associated with poor prognosis for patients’ survival. This demonstrates that ICOS participates in T cell mediated immunosubversion and suggests ICOS neutralization on Treg as a new cancer immunotherapy strategy
8.2.3 Treg-Mediated Suppression
Treg can suppress most immune cells including CD4+ and CD8+ T cells, DC, B cells, MΦ and NK cells. In vivo and in vitro studies suggest Treg-mediated suppression could be operated through multiple mechanisms and that various molecules could be secreted or expressed at cell surface and actively participate simultaneously and synergistically to their suppressive functions on these different cell subsets (Fig. 8.5).
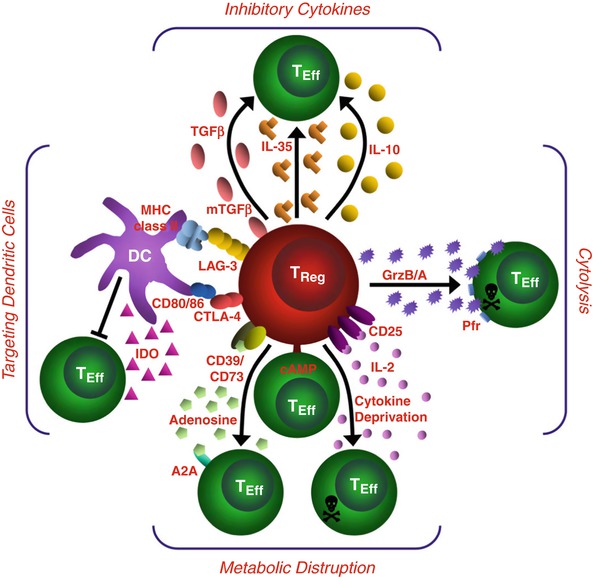
IL2: IL-2 may increase Treg suppressive function by upregulating FOXP3 expression via STAT5. Moreover, Treg could deprive local IL-2 by consumption through their high-affinity IL-2Rα receptor (CD25).
Cytokine secretion: IL-10 and TGF-β contribute to nTreg and iTreg-mediated suppression.
Granzyme-dependent cell cytolysis: Human activated Treg express perforin and granzyme A after activation and can kill activated CD4+ and CD8+ T cells and other cell types independently from Fas/FasL [72–74].
Extracellular nucleotide metabolism via CD39 and CD73: CD73 cooperates with CD39 to generate adenosine (Ado) from ATP [75]. CD39 predominantly catalyzes conversion of ATP into AMP, which in turn is degraded by CD73 to produce Ado. To mediate suppression, Ado binds one of the four distinct AdoR (A1, A2A, A2B, A3). A2A and A2B receptors are coupled to Gs subunit that activates adenylate cyclase and protein kinase A (PKA) thereby increasing cAMP levels. Binding to A2B and A2A suppresses the function of both innate and adaptive immune cells (for review [76]). Indeed, through A2B receptor, Ado alters the maturation of APCs (monocytes and DC) and their secretion of pro-inflammatory cytokines and favors an immunosuppressive environment through induction of IL-10 secretion by monocytes [77]. Furthermore, through A2A receptor, Ado reduces cytotoxic capacity of NK cells [78], decreases IFN-γ secretion, and favors secretion of immunosuppressive cytokines (IL-4 and TGF-β) by NKT cells [79], inhibits BCR-induced NFκB activation [80] and reduces major functionalities of effector CD4+ and CD8+ T cells such as IL-2 and IFNγ secretion, proliferation, and cytolytic function and alters their survival [81]. Together Ado will alter the development of an effective anti-tumor immune response.
The degradation of ATP into Ado by CD39 in tandem with CD73 represents a mechanism that is used by Treg to induce the production of peri-cellular Ado. However, whereas most studies reported CD39 and CD73 co-expression on murine Treg [82–85], our work showed that in human peripheral blood, lymphoid tissues or breast tumor tissues, Treg never expressed CD73 either on the membrane or in the cytoplasm (Gourdin et al. submitted). Similarly to mouse [86], in human breast tumors, Treg express high CD39 levels (Gourdin et al. submitted). Moreover, TLR-7-activated pDC selectively induce the upregulation of CD39 levels on Treg (unpublished data). However, CD73 is observed on a subset of non-Treg CD4+ T cells able to proliferate and secrete high IFN-γ levels upon activation. Our recent data show that CD39+ TATreg cooperate with CD73+CD4+ T cell effectors to mediate Ado production and their local suppressive function (Gourdin et al. submitted) highlighting CD39/CD73 axis as an important Treg-mediated suppression mechanism.
Targeting DC: Interaction of CTLA-4, constitutively expressed on Treg surface with its ligands CD80 and CD86 on DC is an important pathway by which Treg could mediate their suppressive function. Indeed, Treg downregulate or prevent CD80 and CD86 upregulation on murine as well as human DC in vitro blocking their co-stimulatory function and subsequent T cell activation and function [87, 88]. Lag-3 expressed on Treg may play a role in Treg-induced suppression of DC function as Lag-3, by binding MHC class-II molecules expressed by immature DC, induces an ITAM-mediated inhibitory signal that blocks their maturation and reduces their allo-stimulatory functions [89].
Fig. 8.5
Mechanisms of Treg suppression. This schematic depicts the various Treg mechanisms arranged into four basic modes of action. “Inhibitory cytokines” include IL-10, IL-35, and TGF-β. “Cytolysis” includes granzyme-A- and granzyme-B-dependent and perforin-dependent killing mechanisms. “Metabolic disruption” includes high-affinity IL-2 receptor α (CD25)-dependent cytokine-deprivation-mediated apoptosis, cyclic AMP (cAMP)-mediated inhibition, and CD39- and/or CD73-generated, adenosine–purinergic adenosine receptor (A2A)-mediated immunosuppression. “Targeting dendritic cells” include mechanisms that modulate DC maturation and/or function such as LAG3 (also known as CD223)–MHC-class-II-mediated suppression of DC maturation, and CTLA4–CD80/CD86-mediated induction of indoleamine 2,3-dioxygenase (IDO), which is an immunosuppressive enzyme, by DCs
8.3 Therapeutic Strategies
8.3.1 Strategies to Neutralize Treg Function Based on pDC/Treg Interaction
We developed a clinically relevant murine tumor model in which the HER2/neu+ NEU-15 cell line growing in WT hosts escapes from immunosurveillance through pDC and Treg-mediated immunosubversion [63], thus closely mimicking our observations in human breast cancer (Fig. 8.6) [32, 33, 35, 42, 62].
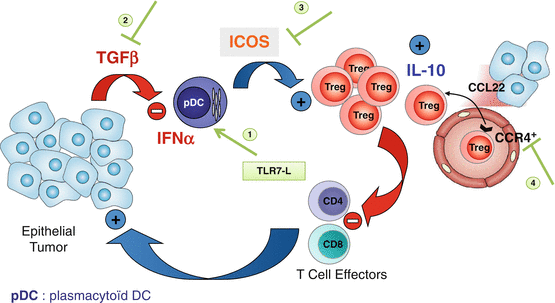
Fig. 8.6
 Peptide-Based Therapeutic Cancer Vaccines
Peptide-Based Therapeutic Cancer Vaccines
 The Human Tumor Microenvironment
The Human Tumor Microenvironment
 PD1 Checkpoint Blockade in Melanoma: From Monotherapy to Combination Therapies
PD1 Checkpoint Blockade in Melanoma: From Monotherapy to Combination Therapies
 Cancer Immunosurveillance by Natural Killer Cells and Other Innate Lymphoid Cells
Cancer Immunosurveillance by Natural Killer Cells and Other Innate Lymphoid Cells
 Immune Checkpoint Inhibition in Lung Cancer
Immune Checkpoint Inhibition in Lung Cancer
 Immune Monitoring of Blood and Tumor Microenvironment
Immune Monitoring of Blood and Tumor Microenvironment




Therapeutic strategies through targeting pDC/Treg crosstalk. Based on the observation of pDC-mediated Treg expansion we are proposing several complementary approaches to reactivate anti-tumor immunity. (1) pDC reactivation for type-I IFN production through TLR-7 ligands that are currently under clinical trials; (2) TGF-β neutralization to restore pDC function, several mAbs against TGF-β receptor or TGF-β or TGF-β receptor kinase domain inhibitors are also under clinical trials; (3) ICOS neutralization though ICOS/ICOSL neutralizing mAbs to block Treg proliferation; (4) mAbs neutralizing CCL22/CCR4 to block Treg recruitment within the tumor, neutralizing anti-CCR4 mAbs or small inhibitors are under clinical trials and/or development
Related posts:
Peptide-Based Therapeutic Cancer Vaccines
The Human Tumor Microenvironment
PD1 Checkpoint Blockade in Melanoma: From Monotherapy to Combination Therapies
Cancer Immunosurveillance by Natural Killer Cells and Other Innate Lymphoid Cells
Immune Checkpoint Inhibition in Lung Cancer
Immune Monitoring of Blood and Tumor Microenvironment
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
