Albumin was the only plasma concentrate used in World War II. However the other plasma fractions were retained and analyzed later. An entire industry which now involves tons of plasma and billions of dollars evolved from these early studies.
The Fractionation Process
Paid donor apheresis plasma (‘source plasma’) is the major source of raw material for the commercial plasma products industry. Plasma is collected from dedicated donors, often paid to donate as often as twice a week. These donors are monitored by a health history, physical examination, and serial laboratory testing. The donation process involves automated instrumentation that draws as much as 800 ml at a sitting, depending upon the size of the donor. Plasma is often drawn into 4% citrate anticoagulant (6.5 ml/100 ml blood processed), frozen quarantined until testing is complete, and shipped to fractionation plants. To a much lesser extent, the commercial industry relies on plasma recovered from whole blood donations (‘recovered plasma’) although this is the major source of supply for some national blood services, particularly those that wish to develop plasma independence based on volunteer, unremunerated donation. Recovered plasma may be mixed with any of the several anticoagulant/preservative solutions used worldwide for whole blood collection and may dilute to rations as high as 25 ml per 100 ml blood collected (for ACD-B). In the US, which supplies the majority of the plasma for commercial fractionation worldwide, plasma fractionation must adhere to Good Manufacturing Practices (GMP) from selection of donors through manufacture and distribution of the finished products (Burnouf 2007). The industry is highly regulated by national regulatory authorities. Plasma donors are screened and tested in a manner analogous to that used for whole blood donor screening. Plasma is labelled and scanned into a system to guarantee traceability, and stored, shipped, and pooled in a manner that ensures maintenance of the ultra-low temperature cold chain. Plasma pools for fractionation range from 10 000–50 000 donations.
The most widely used method of fractionating plasma remains the cold alcohol precipitation technique described by Cohn and colleagues (1944) or modifications thereof (Kistler and Nitschmann 1962). Cohn fractionation relies on changes in ethanol concentration and pH for bulk precipitation of different protein fractions. An example of a fractionation scheme is shown in Figure 19.1. Ethanol is removed by lyophilization or by ultrafiltration. Alcohol fractionation is now combined with glycine precipitation or polyethylene glycol, and with other separation methods such as chromatography to isolate specific proteins, such as coagulation factors and protease inhibitors.
Pools are thawed slowly at 1° to 5°C to produce cryoprecipitate, a cold-insoluble fraction that contains significant amounts of factor VIII, von Willebrand factor (vWF), fibrinogen, fibronectin, and factor XIII, along with a number of other proteins present in smaller quantities. The cryoprecipitate is usually recovered by centrifugation. The cryo-supernatant or cryo-poor plasma may be treated with a chromatographic media to capture the factor IX complex or antithrombin (AT) before it enters the Cohn process. There it goes through a series of precipitations as the ethanol concentration is increased in steps from 8% to 40% at specific combinations of pH, ionic strength, protein concentration, and cold temperature. The precipitates and supernatants are separated either by the traditional continuous-flow centrifugation or in large-scale filter presses. The method provides both relatively pure fractions containing albumin and immunoglobulins, which need minimal additional processing, as well as fractions enriched in other proteins, which are used as the starting materials for further purification. The first precipitate, fraction I at 8% alcohol, contains factor VIII, fibrinogen, and other poorly soluble proteins. Fractions II and III are precipitated together and contain the immunoglobulins, which are separated in a subsequent series of precipitations to produce fraction II, essentially pure immunoglobulins. Because many of the fraction I proteins are removed in the cryoprecipitate, some manufacturers do not produce a separate fraction I. Instead they collect a combined fraction I + II + III. Fraction IV, produced from the supernatant of fraction (I+)II + III, is sometimes produced in two subfractions. Fraction IV-1 contains the vitamin K-dependent (VKD) clotting factors, AT and α1-proteinase inhibitor (API), whereas fraction IV-4 contains transferrin, haptoglobin, and some of the albumin. Fraction V is almost pure albumin.
Recombinant Plasma Proteins
In 1977, scientists at Genentech produced the first human recombinant protein (somatostatin) expressed by Escherichia coli, and subsequently cloned human insulin which became the first recombinant protein to be commercialized in 1982 (Itakura et al. 1977). In the 1980s, on the heels of the AIDS epidemic and the devastation of the haemophilia community, Factor IX (FIX) and Factor VIII (FVIII) gene cloning and expression in mammalian cell culture systems moved from the laboratory to the biotech industry (Kurachi and Davie 1982; Wood et al. 1984). The history of this development has been recounted elsewhere (Gitschier 2004). Producing substantial amounts of complex human proteins with the post-translational modifications required for proper protein folding, glycosylation, and functionality required, and resulted in a number of different expression systems, including yeast, mammalian cells, and transgenic animals (Burnouf 2011). As a result, a variety of genetically engineered therapeutic plasma proteins have now entered the market.
Selected Protein Fractions
Albumin
Albumin, a 66.5 kDa single chain protein, is the most abundant protein in blood plasma. Plasma-derived human albumin for human use derives from Cohn fraction V and is available in saline containing 4%, 4.5%, 5%, 20% or 25% protein, of which not less than 95% is albumin (Table 19.1). Plasma volume replacement is the major indication for albumin infusion (see Chapter 2). A second albumin solution known as plasma protein fraction (PPF), made by co-precipitating fraction IV-4 with fraction V, is available only as a 5% solution of which at least 83% is albumin. Compared with albumin, most preparations of PPF contain larger amounts of excipient proteins (Hink et al. 1957). Hypotensive reactions attributed to pre-kallikrein activator and acetate have been observed with rapid PPF infusion, but not with albumin (Alving et al. 1978; Ng et al. 1981). For these reasons, most clinicians find little reason to select PPF when an albumin solution is indicated.
Table 19.1 Selected human plasma proteins.
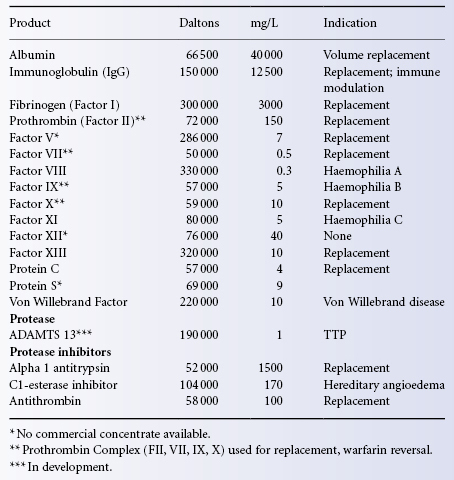
Albumin preparations are filtered and pasteurized by heat treatment at 60°C for 10 h. When prepared in this way, the fraction has proved free of transfusion-transmitted agents such as hepatitis viruses and HIV. In order to prevent denaturation of the protein during pasteurization, the short-chain fatty acids caprylic acid and N-acetyl-DL-tryptophan are added during the manufacturing process. These stabilizers bind tightly to albumin sites that are ordinarily responsible for drug binding and transport (Olsen et al. 2004). The clinical importance of this fractionation step is that more free drug that is ordinarily albumin-bound may be made available during massive albumin infusions or plasma exchange procedures (Reine et al. 2010).
Although albumin contributes 75–80% of the colloid osmotic pressure of the plasma, subjects with a genetically determined total absence of plasma albumin, in whom the colloid osmotic pressure of plasma is between one-third and one-half of normal, may be completely asymptomatic (Bearn 1978). Such subjects show an increase in various plasma globulins and a slight decrease in blood pressure, changes that are regarded as compensatory. The indications for infusions of albumin in hypovolaemic patients are discussed in Chapter 2.
Recombinant Albumin
Recombinant albumin has been synthesized in yeast, in Saccharomyces cerevisiae, or in Pichia pastoris, and appears to be similar to the plasma-derived protein (Dodsworth et al. 1996). However cost effective production on a commercial scale has been challenging. A 20% solution (Recombumin 20%, Aventis Behring) prepared as a pharmaceutical excipient has been tested for safety in doses up to 65 mg in some 500 subjects. In an open-label, parallel-group study of high purity, mass-produced recombinant albumin derived from P. pastoris, pharmacokinetics and bioequivalence were similar to plasma-derived albumin in 22 patients with liver cirrhosis after repeated intravenous infusions at a daily dose of 25 g for 8 days (Ohnishi et al. 2008). Finally, a herd of transgenic cows expressing levels of recombinant albumin as high as 40 g/l in their milk has been created (Echelard et al. 2009). It is uncertain when, if ever, recombinant albumin might be commercially available as a therapeutic product for transfusion.
Fibrinogen Concentrate (Factor I)
Fibrinogen is a dimeric plasma glycoprotein consisting of three pairs of polypeptide chains (Aα, Bβ and γ) with a molecular weight of about 340 kDa. Fibrinogen is cleaved by thrombin to form fibrin monomer (fibrin) which polymerizes to covalently linked fibrin and is crosslinked by factor XIIIa (see below) to form a stable fibrin clot (Hoffman and Monroe 2007).
With the development of component therapy, clinical syndromes that were complicated by hypofibrinogenaemia were managed with infusions of plasma or of cryoprecipitate, which concentrates fibrinogen, factor VIII and factor XIII (see Chapter 14). Early concentrates of fibrinogen prepared by fractionation of large pools of plasma carried an unacceptably high risk of transmitting viral diseases, particularly hepatitis. A commercial source of heat-treated, lyophilized fibrinogen derived from the cryoprecipitate of pooled human plasma and labelled for content (generally about 1 g per vial) is now widely available. The manufacturing process reduces the risk of viral transmission.
The rate of disappearance of injected fibrinogen has been studied by giving infusions to patients with the rare condition, hereditary afibrinogenaemia: in two cases, one-half of the injected fibrinogen disappeared during the first 24 h, presumably due to mixing with the protein ‘pool’; thereafter the fibrinogen disappeared with a T1/2 of 4 days (Gitlin and Borges 1953). The commercial concentrate has a T1/2 of approximately 80 h.
Fibrinogen concentrate is used for treating bleeding episodes in patients with congenital fibrinogen deficiency. Infusions for managing other clinical situations of acquired hypofibrinogenaemia (trauma, cardiac surgery, postpartum haemorrhage, dilutional hypofibringenaemia) are supported by passionate adherents, but meagre evidence (Danés et al. 2008). When administered, the fibrinogen therapeutic dose is generally calculated based on patient weight:
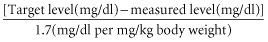
However when fibrinogen level is unknown, a dose of 70 mg/kg is generally used. Venous thrombosis has been reported in patients with congenital afibrinogenemia both with and without infusion therapy (Peyvandi et al. 2006).
Factor VIII (Anti-Haemophilic Factor)
Factor VIII Levels in Haemophiliacs
Haemophilia, the most common, severe inherited bleeding disorder, has been recognized since Biblical times. Not until the 1950s were the two major forms of haemophilia (A and B) distinguished. Severely affected patients with haemophilia A have no detectable factor VIII activity in their plasma and suffer from repeated episodes of spontaneous bleeding, particularly in the large joints and muscles. Factor VIII deficiency is X-linked and occurs in about 1/10 000 male births. Patients whose factor VIII activity is 1–5% of normal, about 10% of affected individuals, are defined as ‘moderate haemophilia’ and have infrequent attacks of bleeding. Those with levels exceeding 5%, some 30–40% of patients, are mildly affected and seldom if ever suffer spontaneous bleeding. Intracranial haemorrhage is the most common cause of death from bleeding, is spontaneous in about 50% of cases and should always be considered in patients with haemophilia who complain of headache.
Factor VIII is a complex heterodimer that is synthesized in the liver as a 300-kD procofactor and circulates bound to its chaperone molecule, von Willebrand factor (vWF). The gene encoding the factor VIII protein is located at Xq28. The protein consists of three types of domains, three A domains, which have a 35% amino acid sequence homology to factor V, a central B domain with no known homologues, and two C-terminal domains that are also 35% homologous to factor V. A comprehensive internet database of haemophilia A mutations is maintained at http://hadb.org.uk.
Treatment with Factor VIII
In severe haemophilia A, treatment with factor VIII for bleeding must be provided as soon as possible to reduce extension of the haemorrhage and prevent chronic disability. Although plasma and cryoprecipitate contain measurable levels of factor VIII, the risks of volume overload and viral transmission have rendered them obsolete except when concentrates are unavailable. The dose depends on the kind of haemorrhage. The aim of initial ‘episodic’ therapy in the case of haemarthrosis or serious bruising is to raise the factor VIII level to 30–50% of normal; if a soft tissue haematoma has developed, the level should be raised to 50%, and in cases of gastrointestinal bleeding, to >50%. The level should be raised to 100% if there is spontaneous intracerebral haemorrhage or head trauma (Furie et al. 1994). One unit of factor VIII is the amount of factor VIII activity in 1 ml of normal plasma. For example, as the plasma volume is about 50 ml/kg, 3500 units must be given to a recipient of 70 kg to increase the level to 100%. Regardless of the source of factor VIII, the plasma level reached after administration is only about 70% of the expected level and this must be taken into account in calculating the dose required. The half-life of factor VIII is 8–12 h. Thus if one-half of a dose is given at 12-h intervals after the initial dose, the level is kept relatively constant (Furie et al. 1994). All currently available plasma-derived and recombinant concentrates have similar pharmacokinetics.
Prophylactic Administration of Factor VIII
The incidence of bleeding can be all but abolished and even arthropathy can be prevented if factor VIII is administered prophylactically from a very early age so as to maintain the factor VIII concentration above 1% of normal (Manco-Johnson et al. 2007). Once joint damage has occurred, it cannot be reversed by prophylactic treatment (Manco-Johnson et al. 1994). Several prophylactic regimens have been proposed, ranging from the full-dose (Malmo) regimen of 25–40 U/kg three times a week, to low-dose prophylaxis, 10–15 U/kg once or twice a week. Most national organizations now recommend primary prophylaxis for children under the age of 2 years with severe haemophilia. In adults, data on the efficacy of long-term prophylaxis, as well as the optimal dose, interval, and measure(s) of efficacy are lacking (Stobart et al. 2010). Short-term prophylaxis (3 months of bi-weekly infusions calculated to keep trough values at 1–3%) should be considered for patients with frequent haemorrhages or with chronic synovitis, especially if active rehabilitation is considered (Kasper et al. 1989). With preoperative prophylaxis, surgical mortality from haemorrhage approaches zero for most procedures (Kitchens 1986). The initial infusion is routinely given several hours prior to surgery and the factor VIII level confirmed before induction of anaesthesia. The duration of postoperative infusions depends on both the nature of the procedure and the clinical course (Kasper et al. 1985). Continuous postoperative factor infusion has become an increasingly popular strategy to maintain constant factor levels (>50%) and consume less factor concentrate, although experience with this technique is still limited.
Factor VIII Levels of Healthy Donors: Maximizing Collection Potential
The factor VIII activity in group A donors is on average 8% higher than in group O donors, and the level in males is about 6% higher than in females (Preston and Barr 1964). Strenuous exercise produces an almost immediate increase in factor VIII levels, lasting for at least 6 h (Rizza 1961). These observations carried greater importance when cryoprecipitate was used as a source of factor VIII.
DDAVP (1-deamino-8-D arginine vasopressin, also known as desmopressin acetate), a synthetic derivative of vasopressin, injected intravenously, produces a rapid release of vWF into the circulation. Although vWF is a carrier protein for factor VIII, factor VIII and vWF may not increase concurrently (Cattaneo et al. 1994; Castaman et al. 1995). DDAVP is the treatment of choice in patients with mild haemophilia with factor VIII levels of >10%, but should not be used in children under 1 year of age because of the risk of hyponatraemia (Weinstein et al. 1989; see also Chapter 18).
DDAVP is used primarily for therapeutic purposes. However, if DDAVP (0.2 µg/kg) is injected into blood donors 15 min before venepuncture, the yield of factor VIII in fractions prepared from the resulting plasma is increased two-fold (Nilsson et al. 1979; Mannucci 1986). DDAVP can also be administered intranasally and is effective within 1 h with minimal side-effects (Mikaelson et al. 1982).
Choice of Factor VIII Concentrate
Factor VIII can be administered as cryoprecipitate (see above and Chapter 14), as plasma-derived factor VIII concentrate or as recombinant factor VIII. Factor VIII activity is well maintained at −30°C to −40°C for cryoprecipitate or at 4°C for lyophilized products. Commercial plasma-derived factor VIII is now treated with at least two methods for inactivating transfusion-transmitted viruses, and no documented transmissions of the lipid-encapsulated viruses HIV, HBV or HCV have been reported since 1987. Non-enveloped viruses such as hepatitis A and parvovirus B19 may still be transmitted by plasma fractions (Mannucci 1992; Santagostino et al. 1997). Both plasma-derived and recombinant products are labeled for factor VIII potency and all preparations appear to be equally effective when assessed by post-administration factor levels. Patient age, susceptibility and product cost still largely determine the choice of treatment.
Purified Factor VIII Concentrates
Plasma-derived concentrates are generally classified as high purity (containing only the desired factor or intermediate purity (containing the desired factor but small amounts of other proteins). In the case of factor VIII, intermediate-purity products generally contain vWF (e.g., Alphanate [Grifols], Humate [CSL-Behring], Wilate [Octapharma]).
Intermediate-/high-Purity Concentrate
Factor purity is commonly defined as specific activity (International Units of clotting activity per milligram of protein). Most fractionation centres use large pools of plasma (5000–30 000 donations) to prepare products of intermediate purity (<50 units/mg) and high purity (>50 units/mg). The primary procedure is cryoprecipitation, but additional fractionation steps are undertaken to give a higher potency, stability and solubility than are obtained with the freeze-dried cryoprecipitate.
Ultrapure Concentrate
Concentrates purified by using affinity chromatography with monoclonal antibodies against factor VIII have a specific activity of factor VIIIC of >3000 IU/mg protein. In a cohort of patients treated with this product for more than 24 months, clinical efficacy, T1/2 and recovery were excellent (Brettler et al. 1989). One oft-stated advantage of purified high-potency concentrates is a less pronounced effect on the immune system of the patients, documented primarily in HIV-positive patients in whom the CD4+ cell count declines less rapidly after treatment with high- than after intermediate-purity concentrates (de Biasi et al. 1991; Hilgartner et al. 1993; Seremetis et al. 1993). No increase in AIDS-associated infections or decrease in survival has been documented (Goedert et al. 1994). No difference in effect on the immune system in HIV-negative patients has been established. Suspicion that ultra high-purity concentrates may more easily induce factor VIII inhibitors, particularly in children, have not been confirmed by prospective studies (Bray 1994; Peerlinck 1994). The relationship between mutation type and inhibitor development is probably far more important. As many as 35% of patients with ‘severe molecular defects’, intron 22 inversions, large deletions or stop mutations develop an inhibitor, whereas few with small insertions or deletions do so (Oldenburg et al. 2002).
Recombinant Factor VIII
Factor VIII is the largest and most complex protein prepared commercially by recombinant technology. Recombinant clones encoding the complete 2351-amino-acid sequence have been used to produce factor VIII in cultured mammalian cells; the first-generation protein was stabilized with human serum albumin (Wood et al. 1984). A B-domain deleted second-generation protein has been produced with dramatically enhanced expression, no requirement for addition of human-derived excipients, and similar haemostatic activit (Brondyk 2009). Additional recombinant concentrates are now available including full-length proteins; Factor VIII is co-expressed with vWF so that it is stable in albumin-free medium (Grillberger et al. 2009). The recombinant proteins correct the clotting time of plasma from haemophiliacs and are functionally indistinguishable from plasma-derived factor VIII.
Clinical trials have shown that in vivo recovery and T1/2 of recombinant factor VIII are not significantly different from those of plasma-derived factor VIII. Recombinate (Baxter Bioscience) and Koginate (Cutter Biological/Miles) have been used successfully in the treatment of bleeding episodes and for prophylaxis. The observed incidence of inhibitor formation is similar to studies of previously untreated patients (PUPs) receiving plasma-derived factor VIII. Long-term trials with the early products demonstrated safety and efficacy of recombinant factor VIII in chronic treatment of haemophilia A (Lusher 1994; White et al. 1997).
Animal Factor VIII Concentrates
Concentrates prepared from bovine or porcine plasma have 100 times more factor VIII activity per milligram of protein than normal human plasma. The original preparations were immunogenic and could as a rule be used effectively for only 7–10 days, following which antibodies against the animal protein developed. A highly purified preparation of porcine factor VIII was developed inthe early 1980s using polyelectrolyte chromatographic fractionation to provide an alternative treatment for patients who had developed factor VIII inhibitors (Giangrande 2012). The polyelectrolyte-fractionated porcine factor VIII concentrate (Hyate : C) appeared to be considerably less antigenic and contained negligible amounts of platelet-aggregating factor (Kernoff et al. 1984).
Factor VIII Inhibitors (Antibodies)
Factor VIII inhibitors are found either as isoantibodies in 15–35% of haemophilia A patients following treatment with factor VIII-containing materials or, more rarely, as autoantibodies in non-haemophiliacs. The titre of the antibody measured in Bethesda units (Bu) determines both risk and management. About one-half of the antibodies in haemophiliacs are of low titre and transient. Patients with factor VIII inhibitors are relatively refractory to treatment with factor VIII and must be given very high doses to secure a response. Concentrates of factor VIII are effective if the inhibitor titre is less than 20 Bu/ml; with higher titres, factor VIII concentrates alone are ineffective. Haemophiliacs with factor VIII inhibitors may show a rise in inhibitor titre following the infusion of factor VIII; in such patients factor VIII concentrates are not effective after 5–7 days of treatment (Blatt 1982). In a cohort study involving 574 consecutive, previously untreated children with severe haemophilia A who were born between 2000 and 2010, recombinant factor VIII products conferred a risk of inhibitor development similar to that of plasma-derived products (Gouw et al. 2013). The rate of inhibitor formation with use of the full-length products may exceed that of the later products. The von Willebrand factor content in factor VIII products was not associated with inhibitor development.
In treating major haemorrhage in haemophiliacs with inhibitors, provided that the inhibitor titre does not exceed 20 Bu/ml, human factor VIII concentrate can be used initially. In average-sized adults (70 kg), 5000 units of factor VIII are given initially, followed by 500–1000 units per hour. Thereafter, the dose is adjusted according to the factor VIII level (Blatt 1982). In patients with inhibitor titres exceeding 20 Bu/ml, activated prothrombin complex concentrates appear to be effective, but have an increased risk of thromboembolic complications (Hilgartner et al. 1990; Tjonnfjord et al. 2004). These concentrates bypass the need for factor VIII in a manner not thoroughly understood. Recombinant activated factor VII (rFVIIa) has been found effective in patients with antibodies against factor VIII (see also Chapter 18) (Hedner and Kisiel 1983; Abshire and Kenet 2004). Standard dosing of rFVIIa (90 µg/kg) allows binding of FVIIa to the surface of activated platelets and can directly activate factor X in the absence of tissue factor. Experience with bolus dosing suggests that higher dosing (>200 µg/kg) may be more efficacious in treating haemophilia patients.
In patients who require very large amounts of factor VIII, animal factor concentrates have been used successfully (Kernoff et al. 1984; Giangrande 2012). Four haemophiliacs with factor VIII inhibitors received repeated infusions of porcine VIII for periods up to 27 days with satisfactory results. An antibody response was detected in only one out of the four patients. Equally good results were obtained in a later trial (Hay et al. 1990). The newer treatments, and the recognition of vCJD prion disease in cattle and other ruminants, should render animal factor concentrates a historical curiosity.
In about 50% of patients with high-titre inhibitors and up to 90% of those with titre <5, immune tolerance can be induced by desensitization regimens that involve daily factor VIII infusions (Ewing et al. 1988). Treatment may be required for weeks or months and infusions of factor VIII are sometimes combined with short courses of immunosuppressive agents such as corticosteroids, cyclophosphamide and IVIG (Kasper et al. 1989; Mariani et al. 1994). The optimum daily dose, immune suppressive regimen, and source of factor VIII have not been defined (Hay and Dimichele 2012).
Treatment with IVIG is beneficial in some patients with haemophilia A and inhibitors and particularly in patients with factor VIII autoantibodies. The effect may be due to anti-idiotype antibodies (Sultan et al. 1994; Schwartz et al. 1995).
Transfusion in Patients with Von Willebrand Disease
Von Willebrand disease is a common inherited autosomal dominant bleeding disorder characterized by easy bruising, epistaxis, bleeding with dental procedures and gastrointestinal haemorrhage. In the majority of patients, bleeding results from decreased vWF, a protein carrier of factor VIII that mediates platelet–platelet interaction and platelet binding to vascular subendothelium (Ruggeri and Ware 1993). The vWF gene is located on chromosome 12, and numerous polymorphisms and mutations have been reported; the von Willebrand syndromes are now classified by molecular, functional and clinical criteria (Sadler 2006).
The VWF protein is a large multimeric glycoprotein (>20 000 kDa) present in plasma in a few mg/l. VWF dimers are produced in endothelium, megakaryocytes, and subendothelial connective tissue and form multimers by crosslinking of cysteine residues. The large multimers exhibit the highest functionality. The mechanism of clearance is largely unknown, but appears to be independent of multimer size.
Appropriate treatment depends on the specific type of vWD. Type 1 vWD, the most common form, is a partial deficiency state and usually presents with mild or moderate bleeding. Laboratory diagnosis shows low vWF antigen, activity (ristocetin cofactor) and factor VIII levels, as well as a prolonged bleeding time. Such patients usually respond well to an infusion of DDAVP (0.3 µg/kg) with release of sufficient vWF and factor VIII from tissue stores within 30 min to elevate these levels several fold for about 4 h (Scott and Montgomery 1993). Some of the less common vWD variants do not respond well to DDAVP (Ruggeri et al. 1980; Sutor 2000). Type 3 vWD is a state of almost complete deficiency. Type 2 disease contains four subtypes, all of which demonstrate different functional abnormalities of the vWF. For the 10–20% of patients with vWD who do not respond to DDAVP and for patients who become refractory (‘tachyphylactic’) with repeated treatment given over a long period of time (Rodeghiero et al. 1992), replacement therapy is available in several forms. Patients who require treatment with vWF should be given high-purity, solvent detergent-treated vWF concentrate (Wilate (Octapharma) are also licensed for the treatment of vWD) (Pasi et al. 1990; Burnouf-Radosevich and Burnouf 1992). If this is not available, factor VIII concentrate which contains vWF should be used instead (Cohen and Kernoff 1990). Alphanate (Grifols USA), a factor VIII concentrate (see above) was the first biologic product approved for treatment of surgical and invasive procedures in patients with vWD in whom the hormone desmopressin is either ineffective or contraindicated. It is not approved for patients with severe vWD (Type 3) who are undergoing major surgery. In clinical studies with Alphanate, 120 major, moderate and minor surgical procedures were performed in 76 patients. More than 90% of patients had favourable outcomes. In these clinical studies, 15.8% of subjects and 5.7% of infusions were accompanied by minor allergic reactions, including itching, facial swelling, rash, and chills. Other preparations including Humate-P (CSL Behring), and Koate-DVI (Talecris Biotherapeutics), have been tested in vitro and in vivo for vWF content and use in therapy for vWD (Agrawal and Dzik 2001). The multimeric pattern of vWF in these concentrates varies, and no product mimics the pattern of multimers that is found in normal human plasma. A recombinant vWF is under development (Turecek et al. 2009).
Cryoprecipitate, although rich in vWF, has generally not been treated to reduce the risk of viral transmission, and is therefore the least desirable of the available components. For planned surgery, the products should be given a few hours before operation and, if factor VIII or ristocetin cofactor is used to estimate effectiveness, the level should be measured immediately before the operation is started; if the level is not high enough (50–100%), more concentrate should be given. Repeated infusions every 12 h may be necessary for a week or more.
Treatment of Factor IX Deficiency (Haemophilia B or Christmas Disease)
Factor IX deficiency is less common than classic haemophilia A and occurs in about 1/30 000 male births. Patients with haemophilia B are treated with factor IX concentrate. The administration of crude factor IX concentrates derived from cryoprecipitate supernatant (‘prothrombin complex concentrate’) was associated with thrombotic complications (Kohler 1999), but highly purified, virus-inactivated factor IX concentrates, based either on a combination of three conventional chromatographic steps (Burnouf et al. 1989) or on immune-affinity chromatography with monoclonal antibodies against factor IX, are now available (Kim et al. 1990; Tharakan et al. 1990). Recombinant factor IX (BeneFIX, Wyeth) produced in a serum-free medium and devoid of stabilizers or human origin is also available. Specific activity ranges from 150–300 IU/mg, and safety and efficacy have been demonstrated in clinical trials (Shapiro et al. 2005). Although the structural and functional characteristics are similar to the plasma-derived protein, there are differences in post-translational glycosylation. Some patients achieve only 80% of the predicted plasma level and interpatient variability is wide when this product is used, so that baseline recovery measurements are important to ensure adequate treatment. Treatment with highly purified or recombinant product avoids the side-effects mentioned above (Kim et al. 1990). Each unit of factor IX administered raises the plasma concentration by 1%. The T1/2 of plasma factor IX is 20 h. As in haemophilia A, the dose needed is determined by the kind of bleeding (see above) (Furie et al. 1994). If no factor IX concentrate is available, PCC (see below) can be used instead. Treatment with PCC may lead to thrombotic disorders and DIC (Aronson and Menache 1987).
Treatment with factor IX concentrate may induce inhibitor formation, although this is less frequent than the formation of factor VIII inhibitors in patients with haemophilia A (Knobel et al. 2002). The treatment of patients with factor IX inhibitors is essentially the same as that of patients with factor VIII inhibitors (see below).
Prothrombin Complex Concentrates (Concentrates Containing Factors II, VII, IX and X, PCCs)
Concentrates containing these vitamin K-dependent factors were originally produced for the treatment of inherited factor IX deficiency but are also used for acquired deficiencies of factors II, VII, IX and X, for example in patients with liver disease or warfarin overdose. Because these four factors have similar structures, they tend to co-purify by most of the methods used to isolate them from plasma. Not all PCCs contain balanced concentrations of the four coagulation factors; some contain minimal amounts of factor VII. Balanced 4 factor PCCs should be chosen as first-line therapy for rapid reversal of warfarin anticoagulation, especially if volume overload is a concern (Holbrook et al. 2012).
As described above, PCCs have also been used for treating minor bleeding episodes in haemophiliacs with inhibitors. One activated prothrombin complex (aPCC) concentrate known as FEIBA (Baxter Bioscience) an acronym for Factor Eight Inhibitor Bypassing Activity, contains activated factors VII, X, and prothrombin in addition to factor IX to enhance thrombin generation by activating the coagulation pathway at points further down than the factor VIIIa/factor IXa step. FEIBA is used most often to curtail active bleeding in patients with high-titre antibodies to factors VIII or IX. However a prospective, randomized, crossover study in haemophilia A patients with high titer inhibitors demonstrated the superiority of prophylactic thrice-weekly FEIBA (85 units/kg) to on-demand therapy in reducing the progression of joint disease (Leissinger et al. 2011). This regimen has been recommended for prophylaxis in patients with high-titre inhibitors to both factors VIII and IX. The concentrates should be administered rapidly, and immediately after reconstitution.
Use of Some Other Coagulation Factors
Factor VII
Fresh-frozen plasma can be used to treat factor VII deficiency; however, the need for frequent infusions and the risk of viral transmission have all but eliminated its use for this indication. Plasma-derived factor VII concentrate has been used successfully to treat patients with hereditary factor VII deficiency (Dike et al. 1980). Long-term prophylaxis in children has also been successful (Cohen et al. 1995). Because of its short half-life (3–4 h), frequent infusions are necessary, especially for surgery, although levels of 15–25% of normal are sufficient for this indication. Recombinant factor VIIa has been found to be effective in haemophiliacs with antibodies to factor VIII and in some subjects with poorly controlled massive or life-threatening haemorrhage (see above and Chapter 18) (Hedner et al. 1988; Schmidt et al. 1994). Recombinant factor VIIa is the treatment of choice when no pathogen-reduced VII concentrate is available (Lusher et al. 1998).
Factor XIII
Factor XIII deficiency, also known as fibrin-stabilizing factor deficiency, is a rare inherited bleeding disorder in which blood clots normally, but the clots formed are unstable, leading to recurrent bleeding (Smith 1990). The protein is thrombin-activated to crosslink fibrin by transamidation and form a stable clot. An estimated one in two million people is afflicted with congenital FXIII deficiency. Bleeding usually presents at birth with continued oozing from the umbilical cord after birth. Poor wound healing and scarring are hallmarks of the deficiency, and miscarriages, joints and muscle haemorrhage following trauma are common. The major risk is intracranial haemorrhage which occurs in 25 to 60% of patients.
Pasteurized factor XIII concentrate is routinely infused prophylactically based on patient weight (40 IU/kg). Because of the long half-life of factor XIII (about 2 weeks), monthly infusions ordinarily suffice and levels above 5% are sufficient to prevent bleeding. Until recently, cryoprecipitate and FFP are have been the components of choice, an remain an effective alternative when the concentrate is not available.
Protein C
Protein C is a serine protease zymogen, which is activated by thrombin to form an endogenous anticoagulant. Activated protein C interferes with the activated forms of factors V and VIII; it requires a cofactor, protein S. Activated protein C also stimulates fibrinolysis by neutralizing the inhibitor of tissue plasminogen activator. Protein C deficiency, whether hereditary or acquired as, for example, in severe liver disease, leads to venous thrombosis. Symptomatic patients with inherited disorders may be homozygous, heterozygous or doubly heterozygous for mutations that result in either deficient or dysfunctional protein. Therapeutic protein C concentrates are now available. A heat-treated protein C concentrate has been used to treat thrombotic episodes, disseminated intravascular coagulation, and purpura fulminans in protein C-deficient patients (Dreyfus et al. 1991). A plasma-derived protein C concentrate prepared by anion exchange chromatography, solvent/detergent treatment, and affinity chromatography has been shown to be safe and effective for long-term therapy in nine patients with inherited PC deficiency, including five infants <5 weeks old and four adults with levels of 14–25% (Dreyfus et al. 2007) and an immunoaffinity-purified, activated protein C concentrate, virus inactivated by chemical treatment (Orthner et al. 1995). Recombinant human activated protein C has anti-inflammatory and profibrinolytic properties in addition to its antithrombotic activity. One recombinant formulation, drotrecogin alpha, produced dose-dependent reductions in the levels of markers of coagulation and inflammation in patients with severe sepsis. In a randomized trial, treatment with drotrecogin alfa activated significantly reduced mortality in patients with severe sepsis, but seemed to be associated with an increased risk of bleeding (Bernard et al. 2001). A Cochran analysis found no evidence to support the use of this drug for treating patients with severe sepsis or septic shock (Martí-Carvajal et al. 2012). The mechanism of the anti-inflammatory effect is poorly understood, but may involve inhibition-n of tumour necrosis factor production by blockade of leucocyte adhesion or interference with thrombin-induced inflammation.
C1 Esterase Inhibitor (C1 INH) and Angioedema
Hereditary angioedema (HAE) was originally reported by Quincke (1882), and the clinical syndrome of recurrent non-pruritic, non-pitting oedema was described in detail by Sir William Osler (1888). HAE is an autosomal dominant disorder caused by mutations in the C1 INH gene and results from a functional deficiency of C1 INH due to either a lack (Type I) or a dysfunction (Type II) of the protein (Donaldson and Evans 1963). More than 200 mutations are known (Pappalardo et al. 2008). A rare form of the clinical syndrome that is not associated with a C1-INH deficiency and is dominantly inherited (HAE type III) has been described in 20 unrelated families, and in six families is associated with two missense mutations in exon 9 of the coagulation factor XII gene (Bork et al. 2009). Recurrent HAE, characterized by swelling of the extremities, face, or tongue, colicky attacks of abdominal pain, and life-threatening laryngeal oedema, can be either hereditary or acquired. Symptoms usually begin in childhood and may recur as frequently as weekly. Attacks may be precipitated by minor trauma, stress or medications such as hormone replacement. Oropharyngeal and dental procedures are a particular risk. The prevalence of this disorder is thought to be between 1/10 000 and 1/50 000 and its penetrance within affected families is variable. Mortality rate in affected kindred approaches 30%, but the severity of the attacks is not related to the C1-INH level. Acquired deficiencies of C1-INH may result from spontaneous mutations or from autoantibodies directed against the protein. This protease inhibitor is involved in the regulation of several proteolytic systems in plasma, including the complement system, the contact system of intrinsic coagulation and kinin release and the fibrinolytic system (Cugno et al. 1990). Functional deficiency of C1-INH permits production of vasoactive peptides including bradykinin that alter vascular permeability. The pathogenesis of HAE is not completely understood (Cugno et al. 2009). Diagnosis is generally confirmed by the finding of a low C4 level, particularly during attacks (Zuraw 2008).
Pasteurized C1-INH concentrates are now available to treat acute attacks of HAE. The swelling responds within hours of treatment, ordinarily 500–1000 U infused intravenously, although larger doses are recommended for laryngeal oedema (20 U/kg) (Gadek et al. 1980; Bork and Barnstedt 2001; Craig et al. 2009, 2010). Because of its long half-life (a range 32–47 hours), this effect is sustained over the duration of an HAE attack (Martinez-Saguer et al. 2010). A recombinant human protein (Rhucin, Pharming) derived from the milk of transgenic rabbits has proved effective as well (50–100 U/kg intravenously) (Zuraw et al. 2010). Acute attacks are also treated with subcutaneous injection of the bradykinin B2 receptor antagonist, icatibant (Lumry et al. 2011). Long-term prophylaxis with C1-INH replacement in hereditary as well as acquired C1-INH deficiency has also been successful (Bork and Witzke 1989; Waytes et al. 1996; Bork and Hardt 2011). Prophylaxis with 17α-alkylated androgen and antfibrinolytic agents is also effective, but no two agents have been compared head-to-head.
Activation of the complement and contact systems occurs in septic shock, together with a decrease of plasma C1-INH. levels. Preliminary results show that complement and contact activation can be diminished by treatment with high-dose C1-INH concentrate (12 000 U) and that survival can be improved (Hack et al. 1992; Igonin et al. 2012). However thrombotic events have been reported in association with C1-INH products when used off-label at high doses. Animal studies have supported a concern about the risk of thrombosis from intravenous administration of C1-INH.
Immunoglobulins
IgG
The relationship between congenital lack of immunoglobulin and recurrent bacterial infection was first reported by Bruton in 1952. Defective antibody production is now recognized as the most common primary immunodeficiency disorder. Patients who lack functional immunoglobulins require life-long replacement therapy to prevent serious bacterial infections. From the early days of cold ethanol fractionation, immunoglobulins were known to concentrate in Cohn fraction II. IgG products are now more often prepared from the supernatant of fraction III or the precipitate II+III for more efficient recovery. Intermediate IgG fractions are subjected to ion-exchange chromatography or PEG precipitation to remove protein contaminants, proteolytic enzymes, and aggregates. Plasma infusion, intramuscular injections, and more recently intravenous and subcutaneous administration of immunoglobulin have all been used as replacement therapy.
Related posts:
 Other red cell antigens
Other red cell antigens
 Red cell antibodies against self-antigens, bound antigens and induced antigens
Red cell antibodies against self-antigens, bound antigens and induced antigens
 Blood grouping techniques
Blood grouping techniques
 Alternatives to allogeneic transfusion
Alternatives to allogeneic transfusion
 The transfusion of platelets, leucocytes, haematopoietic progenitor cells and plasma components
The transfusion of platelets, leucocytes, haematopoietic progenitor cells and plasma components
 The Rh blood group system (including LW and RHAG)
The Rh blood group system (including LW and RHAG)
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


