24 Pituitary Tumors
Epidemiology
Pituitary adenomas are benign neoplasms and comprise the majority of tumors arising in the sella turcica. With approximately 1 in 10,000 people diagnosed annually, these neoplasms are common in the general population.1 According to updated Central Brain Tumor Registry of the United States (CBTRUS) data, pituitary adenomas account for approximately 9% to 11% of all primary intracranial tumor diagnoses.2,3 However, the epidemiology of pituitary adenomas is complicated by the high frequency of small, asymptomatic tumors. Thus, tumor registry data likely underestimate the true prevalence of pituitary tumors. Better estimates of the true prevalence of pituitary adenomas are derived from studies of autopsy specimens or incidental findings on imaging scans. In a recent meta-analysis, the prevalence of pituitary adenomas was determined to be 16.7%, (14.4% from autopsy studies and 22.5% from radiography studies).4 In addition, a recent cross-sectional case-finding survey conducted in a province of Belgium concluded that the prevalence of pituitary adenomas might be even 3 to 5 times higher than previous estimates.5
Anatomy
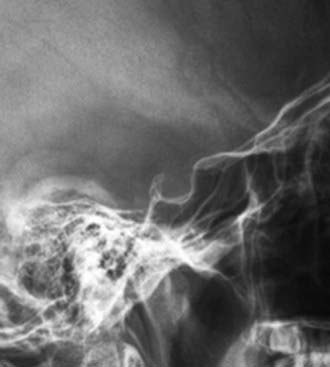
The pituitary gland is situated within the hypophyseal fossa, a fibro-osseous compartment near the center of the cranial base. This fossa is demarcated laterally and superiorly by reflections of dura and elsewhere by the sella turcica (Fig. 24-1), a depression in the body of the sphenoid bone. The diaphragma sellae is an extension of the dura that separates the pituitary from the neural structures located superiorly including the optic chiasm. A central perforation of the diaphragma sellae allows for passage of the infundibulum. The folds of dura mater that form the lateral walls of the hypophyseal fossa contain the cavernous sinuses, which consist of a series of compartmentalized venous channels separated by fibrous trabeculae. The oculomotor nerve, trochlear nerve, and first two divisions of the trigeminal nerve are embedded in the lateral wall of the cavernous sinus, lying between the endothelial lining and the dura mater, whereas the abducens nerve is contained within the sinus itself. The cavernous sinus also envelops a portion of the internal carotid artery and the sympathetic nerve plexus encircling it.
Clinical Presentation
Secretory pituitary tumors present with clinical signs and symptoms attributed to disorders associated with the relevant hypersecreted hormone (Table 24-1). The most common hypersecretory tumor type is prolactinoma. Prolactinomas cause amenorrhea and galactorrhea in women and impotence and infertility in men. The second most common functioning-pituitary tumor is a GH-secreting adenoma which results in acromegaly in adults and in gigantism in children. Cushing’s disease results from the excessive secretion of cortisol as a result of an ACTH-secreting tumor. In the setting of an ACTH-secreting tumor, surgical bilateral adrenalectomy may lead to Nelson’s syndrome, a disorder characterized by aggressive growth of the primary corticotroph adenoma. TSH-secreting adenomas, characterized by thyrotropin-induced hyperthyroidism, are rare. Nonfunctioning pituitary adenomas, arising in the majority of cases from the gonadotropin-producing cells, are not associated with a classical hypersecretory syndrome.
Table 24-1 Clinical Syndromes Associated With Hormonally Functional Pituitary Adenomas
| Hormone Produced | Clinical Syndrome |
|---|---|
| Prolactin (prl) | |
| Growth hormone (GH) | Acromegaly, gigantism |
| Adrenocorticotropic hormone (ACTH) | |
| Thyroid-stimulating hormone (TSH) | Hyperthyroidism |
The modified Hardy classification is sometimes used to describe the tumor size, local growth pattern and extension.6,7 Table 24-2 summarizes the grading of sellar floor destruction and staging of suprasellar or parasellar extension of pituitary adenomas on the basis of radiographic and operative findings.
Table 24-2 Grading of Pituitary Adenomas
| Grading of Sellar Floor Destruction | ||
| Intact sellar floor | I | Sella normal or focally expanded, tumor <10 mm |
| II | Sella enlarged, tumor ≥10 mm | |
| Sellar floor not intact | III | Localized perforation of the sellar floor |
| IV | Diffuse destruction of the sellar floor | |
| V | Spread via cerebrospinal fluid or blood | |
| Staging of Degree of Suprasellar/Parasellar Extension | ||
| 0 | Confined within sella | |
| Suprasellar extension | A | Occupies suprasellar cistern |
| B | Obliteration of the recesses of the third ventricle | |
| C | Gross displacement of the third ventricle | |
| Parasellar extension | D | Intradural extension into anterior, middle, or posterior cranial fossa |
| E | Extension into or beneath cavernous sinus | |
Types of Pituitary Adenomas
Secretory Adenomas
Prolactinoma
Prolactinomas are the most common type of pituitary adenoma, and account for approximately 40% to 45% of all pituitary tumors.8 In young adults, prolactinomas are more common in women, though this gender discrepancy is less apparent in middle-aged subjects. This gender discrepancy may reflect earlier detection in women, as hyperprolactinemia may cause oligo/amenorrhea, galactorrhea, infertility, and androgenization with hirsutism and acne. By contrast, diagnosis is often delayed in men, as the primary symptoms include reduced sexual function and libido.9 The finding of a higher prevalence of macroprolactinomas (>1 cm) and accompanying local mass effects in men may reflect this delay in diagnosis, though a gender effect on prolactinoma growth may be present as prolactinomas in men have higher proliferative indices (e.g., Ki-67) than in women.10,11 In all subjects, indications for therapy include the presence of hypogonadism, bothersome galactorrhea, infertility, headache, and local mass effects including visual field loss. The primary mode of therapy is medical with use of dopamine agonists, which rapidly reduce prolactin secretion and pituitary adenomas size in more than 90% of subjects. Administration of the dopamine agonist bromocriptine, either daily or in split daily doses, results in normalization of serum prolactin or return of ovulatory function in 80% to 90% of subjects.12 Another dopamine agonist, cabergoline, is an ergot-derived dopamine agonist that is administered orally once or twice a week and, in macroprolactinomas, can normalize serum prolactin in 75% and reduce tumor size by about 33% in two thirds of cases.13 Cabergoline may be more efficacious and better tolerated than bromocriptine.14 A potential concern with cabergoline is the finding that use of high doses of cabergoline for Parkinson’s disease may be associated with an increased risk of valvular heart disease,15,16 although the relevance of this finding to prolactinoma management is unclear. Surgery and radiation therapy are indicated in situations where medical therapy fails or the medication is poorly tolerated, or there is a significant cystic component (which does not respond fully to dopamine agonist therapy).17 Because administration of a dopamine agonist at the time of radiosurgery may limit long-term efficacy, it has been suggested that dopamine agonists should be withheld at the time of radiation treatment.18
Acromegaly
Acromegaly is a chronic, debilitating disease characterized by hypersecretion of GH and elevated levels of insulin-like growth factor-1 (IGF-1). This is an uncommon disorder, with prevalence for example in the Newcastle area of Great Britain of approximately 53 individuals per million and an incidence of 3 to 4 new cases per million over 11 years,19 although a recent study from Belgium suggests that the prevalence may be at least five times higher.5 More than 95% of cases are due to a pituitary, somatotroph adenoma, but, in rare cases, acromegaly may be due to neoplasms ectopically producing either GH or GH-releasing hormone.20 Because of the slowly progressive, insidious nature of acromegaly, the diagnosis may be delayed for up to 10 years.21,22 This delay in diagnosis may exaggerate the complications due to the tumor and GH hypersecretion, and is consistent with the finding that the majority of tumors are macroadenomas at detection. Diagnosis and disease control are imperative, as acromegaly is associated with enhanced, premature mortality, and this risk may be negated with biochemical control, including normalization of serum IGF-1 and attainment of safe GH levels.23,24 Acromegaly is also associated with comorbidities including hypertrophic cardiomyopathy, sleep apnea syndrome, arthropathy, colon polyps, carpal tunnel syndrome, along with headaches: biochemical control can significantly improve most of these outcomes as well. Transsphenoidal surgery is the treatment of choice for most patients because it leads to a rapid fall in serum GH levels, is necessary for tumor debulking if there are local mass effects, and, in contrast to medical therapy, may lead to biochemical cure (vs long-term medical “control”).
Approximately 70% to 80% of patients with microadenomas and <50% of patients with macroadenomas attain biochemical normalization after surgery.25,26 Medical therapy is largely utilized as adjuvant therapy for patients who have failed surgery. Somatostatin analogs (octreotide and lanreotide) are available in monthly depot preparations, and control GH and normalize serum IGF-1 levels in approximately 50% to 70% of cases.27 Dopamine agonists, including cabergoline and bromocriptine (both oral agents), are in general less effective than somatostatin analogs.28

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


