26 Pilocytic Astrocytomas and Other Indolent Tumors
By definition, indolent tumors are slow to grow or progress and present with an insidious onset of symptoms over a period of months to years. Generally, such tumors are World Health Organization (WHO) grade I neoplasms with low proliferative potential and may be amenable to cure following resection alone.1 The most prevalent indolent tumor is pilocytic astrocytoma (PA); however, this chapter also discusses other WHO grade I intra-axial neoplasms, categorized as astrocytic tumors, neuronal and mixed neuronal-glial tumors, and ependymal tumors (see Text Box), focusing first on general considerations in therapeutic management, and then on specific considerations relevant to individual tumor subgroups.
Indolent, WHO Grade 1, Intra-Axial Brain Tumors Included in This Chapter
• Astrocytic Tumors
 Pilocytic astrocytoma (PA)
Pilocytic astrocytoma (PA)
 Subependymal giant cell astrocytoma (SEGA)
Subependymal giant cell astrocytoma (SEGA)
• Neuronal or mixed neuronal-glial tumors
 Ganglioglioma and gangliocytoma
Ganglioglioma and gangliocytoma
 Desmoplastic infantile ganglioglioma (DIG) and astrocytoma (DIA)
Desmoplastic infantile ganglioglioma (DIG) and astrocytoma (DIA)
 Dysembryoplastic neuroepithelial tumor (DNT)
Dysembryoplastic neuroepithelial tumor (DNT)
 Papillary glioneuronal tumor
Papillary glioneuronal tumor
 Lhermitte-Duclos disease (dysplastic gangliocytoma of the cerebellum)
Lhermitte-Duclos disease (dysplastic gangliocytoma of the cerebellum)
• Ependymal tumors
 Subependymoma
Subependymoma
Researchers have identified patient populations that are at risk of developing some of these tumors,2–4 and increasingly, genetic mutations associated with these tumors are being identified.
Pearl
• Some patient populations, such as those with neurofibromatosis type 1 (NF1), tuberous sclerosis complex (TSC), and Cowden disease, are at particular risk of developing indolent tumors.
Special Consideration
• Increasingly, researchers are discovering molecular alterations associated with each of these tumors, which is providing a basis for logical strategies in molecularly targeted therapy.
 General Management Considerations
General Management Considerations
Because each neoplasm has unique characteristics and the mode of presentation can vary substantially, the evaluation and treatment of these patients is often individualized. Some lesions present with subtle neurologic symptoms, others with seizures, and still others are incidentally detected. Magnetic resonance imaging (MRI) with and without gadolinium contrast enhancement is the imaging modality of choice to define the location and growth characteristics of the tumor and to guide subsequent management.
A fundamental determination is whether surgery is warranted in a given case and, if so, the timing of the operative intervention. Children with relatively large lesions and significant local mass effect who are minimally symptomatic are scheduled for surgery on the next available operating day, and corticosteroids are generally begun upon diagnosis. In contrast, smaller lesions presenting with seizures and minimal or no mass effect are treated electively or, in some cases, monitored closely with serial imaging, particularly if the growth course of the tumor is uncertain and the lesion location presents a high risk of surgical morbidity. This expectant management approach may also be appropriate for small, indolent lesions that are identified incidentally. For small lesions that are considered to be appropriate for removal, corticosteroids, if warranted, are deferred until surgery, and, in such cases, are typically tapered over 3 to 7 days if significant tumor debulking has been achieved.
Pearl
• If resection is felt to be warranted, the major predictor for a favorable oncological outcome is achieving GTR of the indolent tumor.
• Indolent tumors are frequently associated with epilepsy, so treatment results are measured using both oncology and epilepsy outcomes, and approaches for optimizing seizure control, such as cortical mapping and electrocorticography, may be incorporated within the operative plan.
In treating indolent tumors, the primary predictor of favorable oncological outcome is achieving gross total resection (GTR) of the tumor.5,6 If the initial operation was undertaken with the goal of achieving a GTR and postoperative imaging reveals residual tumor, another attempt at GTR may be a reasonable option before considering adjuvant therapeutic modalities. Additionally, because many indolent tumors are also associated with epilepsy, outcomes are also measured by improvement in seizure control, and multiple preoperative and operative adjuncts may assist with this.7
Multiple surgical adjuncts can assist with preoperative and intraoperative planning. Stereotactic guidance systems allow the surgeon to plan an operative approach that minimizes manipulation of eloquent brain and optimizes the extent of maximum safe resection; however, the limitations of this technology must be recognized.
Cortical stimulation techniques,8 which may be applied extraoperatively, using previously inserted grid or strip electrodes, or intraoperatively, at the time of the planned tumor resection, are useful for identifying speech and motor areas. Additionally, functional MRI and diffusion tensor imaging can localize critical cortical and subcortical areas and pathways to assist with surgical planning.9,10 These functional studies can be fused with stereotactic guidance imaging to precisely delineate relevant loci around the tumor. In patients with intractable epilepsy in association with cortical lesions, electrocorticography can discriminate whether the seizures originate from the lesion alone or if there is additional epileptogenic cortex.8
Pitfall
• Stereotactic guidance systems are affected by brain shift during surgery, so ultrasound and intraoperative MRI have been increasingly used to provide real-time feedback on the location of the lesion and extent of resection.
 Astrocytic Tumors
Astrocytic Tumors
Pilocytic Astrocytomas
Pilocytic astrocytomas are WHO grade I neoplasms accounting for 5 to 6% of all gliomas, primarily affecting children and young adults with no gender predilection.1 They are the most common type of glioma in children, and although they can be located anywhere in the central nervous system (CNS), most are located in the cerebellum (67%).11 Presenting symptoms typically result from tumor mass effect causing focal neurologic signs or seizures. Some patients have nonlocalizing signs due to raised intracranial pressure or hydrocephalus.12 On imaging, PAs are infrequently calcified and often exhibit well-defined borders with enhancement of a mural nodule or a uniform, ring-like pattern (Fig. 26.1).13 In rare cases, such as optic pathway glioma in patients with NF1, in whom such tumors are relatively common, the MRI appearance is sufficient to establish the diagnosis without requiring surgical biopsy. PAs associated with NF1 are considered to be even more indolent than sporadic PAs.2
Special Consideration
• Pilocytic astrocytomas characteristically exhibit alterations in the BRAF gene, most commonly involving translocations between BRAF and KIAA, or activating mutations, such as BRAFv600E, which leads to dysregulated signaling via the mitogen-activated protein kinase (MAPK) pathway.
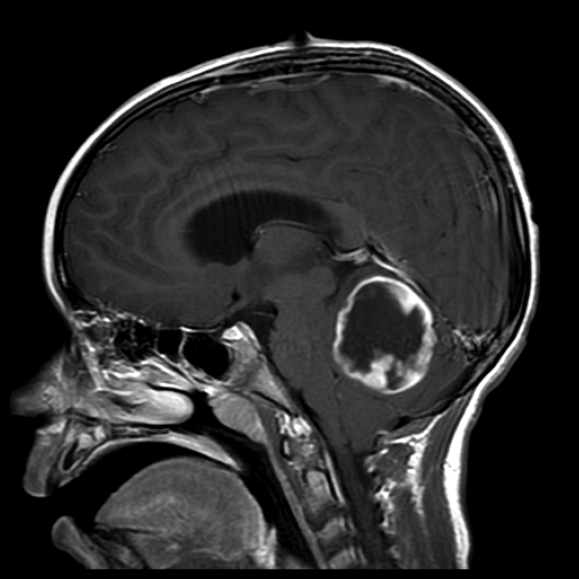
Fig. 26.1 A 7-year-old boy presenting with severe headaches, nausea, and vomiting due to hydrocephalus was found to have this posterior fossa tumor. This sagittal T1-weighted contrast-enhanced magnetic resonance imaging (MRI) scan reveals a cerebellar pilocytic astrocytoma with a mural nodule and enhancing cyst capsule. He underwent suboccipital craniotomy for resection of the nodule and enhancing cyst wall.
• With PAs resection of a nonenhancing cyst wall is unnecessary. In contrast, when the cyst wall is thick and enhancing, resection of the cyst wall is warranted.
Controversy
• Decisions regarding adjuvant therapy are considerably more controversial after a subtotal resection (STR).
Histologically, regions with compact bipolar astrocytes are interspersed with loosely packed multipolar cells containing microcysts. Eosinophilic granular bodies and Rosenthal fibers are also seen. Although not believed to adversely affect prognosis, occasional mitotic figures, leptomeningeal infiltration, vascular proliferation, and necrosis may be noted.1 Recently, PAs were noted to characteristically demonstrate changes in the BRAF gene.14–16
The observation of recurring abnormalities in BRAF or other elements in its signaling pathway has provided insight into strategies for molecularly targeted therapies for PAs. There are ongoing clinical trials of MEK inhibitors, RAF/multiple tyrosine kinase inhibitors such as Sorafenib, and mammalian target of rapamycin (mTOR) inhibitors for tumors not amenable to complete resection or those recurring after resection or initial adjuvant chemotherapy or radiotherapy.17
Because PAs are generally well circumscribed, they are amenable to GTR. The extent of resection has correlated closely with outcome.5,6 Generally, the cyst wall does not need to be excised unless it is enhancing.18 Adjuvant therapy is usually not employed after a GTR because recurrences are infrequent.
Even though at least half of patients after STR will progress within 5 years, overall survival rates exceed 90%.5,6 The observation that PAs may remain quiescent after an incomplete resection suggests that tumors can exhibit decelerated growth kinetics over time, which fits with observations that tumor cells with BRAF alterations may undergo senescence after an initial period of growth.19 Because of this biological variability, many neuro-oncologists prefer to expectantly follow patients with small amounts of residual tumor, only administering additional therapy in the event of tumor progression. In such cases, reoperation for GTR is sometimes feasible6,20,21; if not, adjuvant therapy, including chemotherapy or radiotherapy (RT), may then be employed. Typically, RT is reserved for older children or those who have failed chemotherapy because of the adverse effects of radiation on the developing brain.22,23 A variety of agents and regimens have been observed to delay tumor regrowth and delay or avoid the need for radiation, including carboplatin/vincristine, 6-thioguanine/procarbazine/chloroethylcyclohexylnitrosourea (CCNU)/vincristine, and vin blastine, among others.
Special Consideration
• As a strategy to delay or avoid the need for irradiation, chemotherapy is most frequently used for younger children, who are at increased risk for radiation-related side effects.
Pitfall
• Previously classified as PAs, pilomyxoid astrocytomas are more aggressive and are now classified as WHO grade II.
An attempt at determining the role of RT for incompletely resected low-grade gliomas in the Children’s Cancer Group9891/Pediatric Oncology Group-8930 study failed because of difficulties in patient recruitment. Nonrandomized single-center series suggest that although irradiation significantly increases progression-free survival after incomplete resection, there is no significant impact on overall survival, in part attributed to their increased likelihood of developing malignant lesions within the treatment fields.6,24 Later studies have focused on conformal irradiation with narrow peritumoral margins using three-dimensional image-based treatment planning.25
Rarely, PAs can seed the neuraxis, typically from tumors in the hypothalamic-chiasmatic location.26 PAs have been reported to undergo malignant transformation; however, it is unclear whether this is secondary to radiation-induced changes.24
Pilomyxoid astrocytomas (PMAs) were once classified as PAs, but they are more aggressive tumors, classified as WHO grade II.1 They typically affect infants with a median age at presentation of 10 months. They are most commonly located in the hypothalamic-chiasmatic region.27 Histologically, there is a mucoid matrix with monomorphous bipolar cells radiating around vessels. Ordinarily, there are no Rosenthal fibers or eosinophilic granular bodies.1 These tumors are more aggressive than PAs, with more frequent local recurrence and cerebrospinal spread.28
Subependymal Giant Cell Astrocytomas
Subependymal giant cell astrocytomas are WHO grade I tumors, arising near the midline in close proximity to the foramen of Monro, and develop in 5 to 15% of patients with TSC.29,30 Most commonly, SEGAs develop in the first two decades of life with a mean age at presentation of 11 years.31
Due to their deep location and proximity to the foramen of Monro, patients often develop symptoms from either hydrocephalus or direct compression of the deep nuclei.32 With the increasing use of MRI for screening children with TSC, such tumors are often diagnosed before symptoms develop. On imaging, SEGAs can be difficult to differentiate from subependymal nodules, but if a lesion is larger than 12 mm, enhances, and is near the foramen of Monro, it is likely to be a SEGA (Fig. 26.2).33

Histologically, the tumors consist of large cells resembling astrocytes and are often calcified. Nuclear pleomorphism and multinucleated cells are often present, and there can be increased mitotic activity. Immunoreactivity for both glial and neuronal markers may be noted.1 On molecular analysis, dysregulation of mTOR signaling has been found to underlie the development of SEGAs in TSC.34,35
Surgery has been first-line therapy for SEGAs, which is curative with GTR.33 Typically, a transcallosal or transcortical approach is used for tumor removal; however, purely endoscopic approaches also have been described.36 When complete resection is not feasible, patients typically experience slow growth of the residual tumor, necessitating additional treatment.37 Stereotactic single-dose RT has been used in both primary and adjuvant treatments, but the outcomes have not been consistently positive.38,39 Recent advances in pharmacological therapy for SEGAs have taken advantage of mTOR pathway inhibition, using rapamycin (sirolimus), its prodrug CCI-779 (temsirolimus), or its analogue RAD001 (everolimus) to halt the uncontrolled mTOR pathway.31
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


