Physical Factors
Mats Ljungman
INTRODUCTION
Ionizing radiation (IR) and ultraviolet (UV) light have challenged the genetic integrity of all living organisms throughout time. By inducing DNA damage and subsequent mutations, these physical agents have promoted diversity through natural selection, and, as a result, organisms from all kingdoms of life carry genes that encode proteins that repair damaged DNA. In higher, multicellular organisms, many additional mechanisms of genome preservation have evolved, such as cell cycle checkpoints and apoptosis. Despite the many sophisticated mechanisms to safeguard the human genome from the mutagenic actions of DNA-damaging agents, not all exposed cells successfully restore the integrity of their DNA and some cells may subsequently progress into malignant cancer cells. Furthermore, through manmade activities, we are now exposed to many new physical agents, such as radiofrequency and microwave radiation, electromagnetic fields, asbestos, and nanoparticles, for which evolution has not yet had time to deliver genome-preserving response mechanisms. This chapter will highlight the molecular mechanisms by which these physical agents affect cells and how human exposure may lead to cancer.
IONIZING RADIATION
IR is defined as radiation that has sufficient energy to ionize molecules by displacing electrons from atoms. IR can be electromagnetic, such as x-rays and gamma rays, or can consist of particles, such as electrons, protons, neutrons, alpha particles, or carbon ions. Natural sources of IR make up about 80% of human exposure and medical sources make up about 20%.1 The increased medical use of diagnostic x-rays and computed tomography (CT) scanning procedures likely translates into higher incidences of cancer. Of the natural sources, radon exposure is the most significant exposure risk to humans. Importantly, with better and more comprehensive screening techniques, the human exposure to radon could be dramatically lowered.
Mechanisms of Damage Induction
Linear Energy Transfer
The biologic effects of IR are unique in that the induced damage is clustered due to the local deposition of energy in radiation tracks. The distance between the depositions of energy is biologically very relevant and unique to the energy and the type of radiation. The term linear energy transfer (LET) denotes the energy transferred per unit length of a track of radiation. Electromagnetic radiation, such as x-rays or gamma rays, are sparsely ionizing and therefore classified as low LET radiation, whereas particulate radiation, such as neutrons, protons, and alpha particles, are examples of high LET radiation.1
Radiation Biochemistry
Radiation-induced damage to cellular target molecules, such as DNA, proteins, and lipids, can be either direct or indirect (Fig. 8.1). The direct action of radiation, which is the dominant mode of action of high LET radiation, is due to the deposition of energy directly to the target molecule, resulting in one or more ionization events. The indirect action of radiation is due to the radiolysis of water molecules, which, after initial absorption of radiation energy, become excited and generate different types of radiolysis products where the reactive hydroxyl radical (•OH), can damage both DNA and proteins. About two-thirds of the damage induced by low LET radiation is due to the indirect action of radiation. Since the hydroxyl radical is very reactive (half-life is 10-9 seconds), it does not diffuse more than a few nanometers after it is formed before it reacts with other molecules, and, thus, only radicals formed in close proximity to the target molecule will contribute to the damage of that target.2 However, by chemical recombination of the primary radiolysis products, hydrogen peroxide (H2O2) is formed, which in turn can produce hydroxyl radicals at a later time through the Fenton reaction, involving free metals. Because H2O2 is not very reactive, it can diffuse long distances away from the initial site of energy deposition.
Radical scavengers normally present in cells, such as glutathione, can protect target molecules by reacting with the hydroxyl radical (see Fig. 8.1). Even after the target molecule has been hit and ionized, glutathione can contribute to cell protection by donating a hydrogen atom to the radical, allowing the unpaired electron present in the radical to pair up with the electron from the hydrogen atom. This is considered the simplest of all types of repair and is called chemical repair.3 However, if oxygen molecules are present, they will compete with scavenger molecules for the ionized molecule, and if oxygen reacts with the ionized target molecule before the hydrogen donation occurs, the damage will be solidified as a peroxide, which is not amendable to chemical repair. Instead, this lesion will require enzymatic repair for the restoration of DNA. This augmenting biologic effect of oxygen is called the oxygen effect and is considered an important factor for the effectiveness of radiation therapy.1
Damage to DNA
The direct and indirect effects of radiation induce more or less identical types of lesions in DNA. However, the density of lesions induced in a stretch of DNA is higher for high LET radiation, and this increased complexity is thought to complicate the repair of these lesions. Radiation-induced lesions consist of more than 100 chemically distinct base lesions, such as the mutagenic lesions thymine glycol and 8-hydroxyguanine.2,4,5 Furthermore, damage to the sugar moiety in the backbone of DNA and some types of base damage can result in single-strand breaks (SSB). Because the energy deposition of radiation is clustered even for low LET radiation, it is possible that two individual strand breaks are formed in close proximity on opposite strands, resulting in the formation of a double-strand break (DSB). It has been estimated that 1 Gy of ionizing radiation gives rise to about 40 DSBs, 1,000 SSBs, 1,000 base lesions, and 150 DNA-protein cross-links per cell.2 For a similarly lethal dose of UV light, about 400,000 lesions are required, demonstrating that the lesions induced by IR are much more toxic
than lesions induced by UV light. It is believed that DSBs are the critical lesions that lead to cell lethality following exposure to ionizing radiation.6
than lesions induced by UV light. It is believed that DSBs are the critical lesions that lead to cell lethality following exposure to ionizing radiation.6
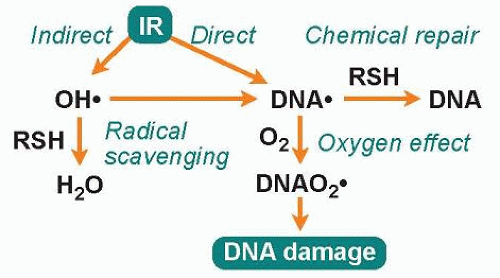 Figure 8.1 Factors affecting the induction of DNA damage by ionizing radiation (IR). Ionizing radiation can ionize DNA either by direct action or by indirect action, in which radiation energy is absorbed by neighboring molecules, such as water, leading to the generation of hydroxyl radicals that attack DNA. Sulfur-containing cellular molecules (RSH), such as glutathione, can scavenge hydroxyl radicals by hydrogen atom donations and thereby protect the DNA from the indirect action of radiation. Glutathione can also donate hydrogen atoms to ionized DNA, thereby restoring the integrity of DNA in a process termed chemical repair. Oxygen can compete with chemical repair in a process termed the oxygen effect, resulting in the enhancement of the biologic effect of ionizing radiation by the fixation of the initial DNA damage into DNA peroxides (DNAO2•). |
Damage to Proteins
Although proteins and lipids are subject to damage following exposure to IR, the common belief is that DNA is the critical target for the biologic effects of radiation. Indeed, abrogation of DNA damage surveillance or repair processes in cells results in the enhanced induction of mutations and decreased cell survival following radiation.5 However, studies of radiation-sensitive and radiation-resistant bacteria imply that mechanisms that suppress protein damage may also play important roles in radiation resistance.7 Deinococcus radiodurans is a bacterium that can survive radiation exposures of up to 17,000 Gy, and its extreme radioresistance has been linked to high intracellular levels of manganese, which protect proteins from oxidation. The thought is that if a cell can limit protein oxidation, then its enzymes will remain active, and cellular functions such as DNA repair will be able to restore the integrity of DNA even after severe DNA damage.8 It would be interesting to explore whether the concentration of manganese can be manipulated to sensitize tumor cells to radiation therapy. Furthermore, because protein damage due to reactive oxygen species (ROS) accumulate during the aging process, could supplements of manganese turn back the clock on aging?
Cellular Responses
DNA Repair
Ever since organisms started to utilize atmospheric oxygen for metabolic respiration many millions of years ago, they have been forced to deal with the cellular damage induced by ROS. Base excision repair (BER) evolved to remove many of the different types of oxidative base lesions and DNA SSBs induced by ROS. However, ROS seldom induce DSBs unless the generation of hydroxyl radicals is clustered near the DNA molecule. A more important source of intracellular generation of DSBs may instead be the process of DNA replication, and it is possible that homologous recombination (HR) repair primarily evolved to overcome DSBs sporadically induced during the replication process. The other major pathway of DSB repair is the nonhomologous end-joining (NHEJ) pathway, which is utilized by immune cells in the process of antibody generation. Although the HR pathway has high fidelity due to the utilization of homologous sister chromatids to ensure that correct DNA ends are joined, the NHEJ pathway lacks this control mechanism and therefore occasionally rejoins ends incorrectly. Thus, the NHEJ pathway may contribute to the generation of mutations following radiation (Fig. 8.2). However, NHEJ is the only mechanism available for DSB repair in postmitotic cells and cells in the G1 phase of the cell cycle because no sister chromatids are available in these cells to support HR repair.
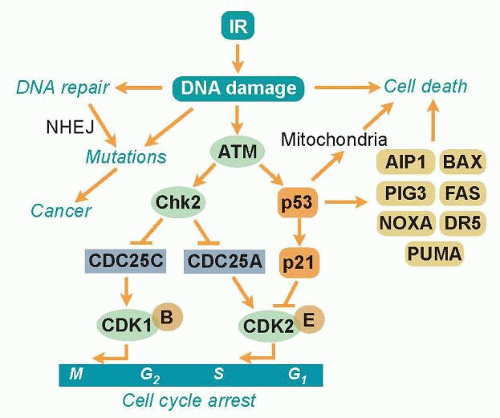 Figure 8.2 Cellular responses to ionizing radiation. Ionizing radiation induces predominantly base lesions and single-and double-strand breaks. Base lesions and single-strand breaks are repaired by base excision repair (BER), whereas double-strand breaks are repaired by nonhomologous end joining (NHEJ) and homologous recombination (HR). If DNA lesions are misrepaired by NHEJ or not repaired at all before cells enter S phase or mitosis, genomic instability is manifested as mutations or chromosome aberrations that promote carcinogenesis. In order for cells to assist DNA repair and safeguard against genetic instability and cancer, cells can induce cell cycle arrest or apoptosis. The ATM kinase is an early responder to DNA damage induced by ionizing radiation that activates the cell cycle checkpoint kinase Chk2 and the tumor suppressor p53. Chk2 inactivates the CDC25A and CDC25C phosphatases that are critical in promoting cell cycle progression by activating the cyclindependent kinases CDK2 or CDK1 and thereby arresting the cells at the G1/S or G2/M checkpoints. In addition, p53 can arrest cells at the G1/S checkpoint by inducing the CDK inhibitor p21. p53 also plays a role in promoting apoptosis by inducing a number of proapoptotic proteins as well as translocating to mitochondria where it inhibits the actions of antiapoptotic factors. AIP1, actin interacting protein 1; BAX, bcl-2-like protein; PIG3, p53-inducible gene 3. |
Ataxia-Telangiectasia Mutated and Cell Cycle Checkpoints
Due to the enormous task of replicating the whole genome during the S phase and segregating the chromosomes during mitosis, proliferating cells are generally much more vulnerable to radiation than stationary cells. To prevent cells with damaged DNA from entering into these critical stages of the cell cycle, cells can activate cell cycle checkpoints (see Fig. 8.2). The major sensor of radiation-induced damage in cells is the ataxia-telangiectasia mutated (ATM) kinase, which, following activation, can phosphorylate more than 700 proteins in cells.9 Two ATM substrates, p53 and Chk2, are critical for the activation of cell cycle arrests at multiple sites in the cell cycle.10,11 The kinase p53 regulates the gene expression of specific genes such as p21, which inhibits cyclin-dependent kinase (CDK)2-and CDK4-mediated phosphorylation of the retinoblastoma protein, resulting in a block in the progression from the G1 phase to the S phase of the cell cycle.12,13 The Chk2 kinase promotes checkpoint activation in G1 by targeting the cell division cycle 25 homolog A (CDC25A) phosphatase14 and, in G2/M, by targeting the CDC25C phosphatase.15 The activation of a cell cycle arrest following DNA damage provides the cell with additional time to repair the DNA before entering critical cell cycle stages,
which promotes genetic stability. Loss or defects in the ATM or p53 genes result in abrogation of radiation-induced cell cycle checkpoints, which manifests itself as the highly cancer-prone human syndromes ataxia telangiectasia16 or Li-Fraumeni,17 respectively.
which promotes genetic stability. Loss or defects in the ATM or p53 genes result in abrogation of radiation-induced cell cycle checkpoints, which manifests itself as the highly cancer-prone human syndromes ataxia telangiectasia16 or Li-Fraumeni,17 respectively.
Radiation-Induced Cell Death
Terminally differentiated and stationary cells, such as kidney, lung, brain, muscle, and liver cells, are generally more resistant to radiation-induced killing than are cells with a high turnover rate, such as different epithelial cells, spermatogonia, and hair follicles. However, the spleen and thymus, which consist of mostly nondividing cells, are among the most radiosensitive tissues, implying that the rate of cell proliferation is not the sole determiner of the radiation sensitivity of a tissue. An important factor regulating the induction of programmed cell death (apoptosis) in tissues is the tumor suppressor p53.18 The p53 protein is activated in cells following exposure to IR by the ATM kinase (see Fig. 8.2). When activated, it regulates the expression of multiple genes that have roles in DNA repair, cell cycle arrest, and apoptosis. p53 can also localize to mitochondria following irradiation, where it triggers apoptosis through the inactivation of antiapoptotic regulatory proteins.19 Not all tissues induce the p53 response to the same degree after similar doses of IR, nor do they activate downstream pathways, such as DNA repair, cell cycle arrest, and apoptosis, in a similar way. For example, thymocytes have an intrinsic setting that favors apoptosis over cell cycle arrest following IR, whereas fibroblasts rarely induce apoptosis, but instead activate a strong and lasting cell cycle arrest.18
IR can induce cell death in tissues by many different mechanisms. Apoptosis can occur rapidly in a p53-dependent manner or later in a p53-independent manner. This later wave of radiationinduced apoptosis is often initiated by mitotic catastrophe, which occurs as a result of complications during chromosome segregation. Cell death induced by IR may in some cases be associated with autophagy, also called autophagocytosis, in which cells degrade cellular components via the lysosomal machinery. Whether autophagy is a programmed cell death or occurs in parallel with cell death is not clear. Interestingly, for some cell types, autophagy has been shown to actually protect the cells from radiationinduced death. Finally, tissue can undergo necrotic cell death following exposure to IR. Necrosis is a clinical problem following radiation therapy that can occur in normal tissues many months after treatment and can contribute to the inflammatory response.
Cancer Risks
It is clear from epidemiologic studies of radiation workers and atomic bomb and Chernobyl victims that IR can induce cancer.20 Twenty years after the atomic bomb explosions in Japan during World War II, significant increases in the incidence of thyroid cancer and leukemia were observed. However, it took almost 50 years before solid tumors appeared in the population as a result of radiation exposure from the atomic bombs.21 The incidences of solid tumors, such as breast, ovary, bladder, lung, and colon cancers, were estimated to have increased by a factor of 2 in the exposed group during this time period. The epidemiology studies following the nuclear power plant disaster in Chernobyl showed a clear increase in thyroid cancer as early as 4 years after the accident.22 Young children were the most vulnerable to radiation exposure, with 1-year-old children being 237-fold more susceptible to thyroid cancer than the control group, while 10-year-old children were found to be sixfold more susceptible to thyroid cancer. Many of the thyroid cancers that developed following the Chernobyl disaster could have been prevented if the population had not consumed locally produced milk that was contaminated with radioactive iodine.
The molecular signatures of radiation-induced tumors are complex but involve point mutations that could lead to the activation of the RAS oncogene or inactivation of the tumor suppressor gene p53. Furthermore, IR induces DNA DSBs that may be unfaithfully repaired by the NHEJ pathway, leading to chromosome rearrangements. One such rearrangement found in 50% to 90% of the thyroid cancers examined following the Chernobyl accident involved the receptor tyrosine kinase c-RET, which promotes cell growth when activated.22 Furthermore, a great majority of the thyroid cancers found in the exposed children harbored kinase fusion oncogenes affecting the mitogen-activated protein kinase (MAPK) signaling pathway.23
The correlation between high exposure to IR and cancer following the atomic bomb explosions and the Chernobyl accident is clear. What about the cancer risk following lower radiation exposures occurring in daily life? There are four theoretical risk models of radiation-induced cancer to consider. First, the linear, no threshold (LNT) model suggests that the induction of cancer is directly proportional to the dose of radiation, even at low doses of exposure. Second, the sublinear or threshold model suggests that below a certain threshold dose the risk of radiation-induced cancers is negligible. At these lower doses of radiation exposure, the DNA damage surveillance and repair mechanisms are thought to be fully capable of safeguarding the DNA to avoid the induction of mutations and cancer. Third, the supralinear or stealth model suggests that doses below a certain threshold or radiation with sufficiently low dose rates may not trigger the activation of DNA damage surveillance and repair mechanisms, resulting in suboptimal activation of cell cycle checkpoints and repair. This would be expected to lead to a higher rate of mutations and cancers than predicted by the LNT model, but may be balanced by a higher incident of cell death. Fourth, the linear-quadratic model suggest that radiation effects at low doses are due to a single track of radiation hitting multiple targets, resulting in a linear induction rate, whereas at higher doses, multiple radiation tracks hit multiple cellular targets, resulting in a quadratic induction rate.
The Biological Effects of Ionizing Radiation (BEIR) VII report, released by the Committee on Biological Effects of Ionizing Radiation of the National Academy of Sciences and commissioned by the US Environmental Protection Agency (EPA), is a review of published data regarding human health and cancer risks from exposure to low levels of IR. Although this topic is controversial and not fully settled, the BIER VII report favored the LNT model.24 Thus, the “official” view is that no level of radiation is safe; therefore, a careful consideration of risks versus benefits is necessary to ensure that the general population only receives radiation doses as low as reasonably achievable. Furthermore, the BIER VII committee concluded that the heritable effects of radiation were not evident in the published data, indicating that an individual is not likely to develop cancer due to radiation exposure of his or her parents.
The largest source of radiation exposure to the population is radon, which is a natural radioactive gas formed as a decay product of radium in the decay chain of uranium. Radon gas can accumulate to high levels in poorly ventilated basements in houses built on rock containing uranium. The major risk with radon is that some of its radioactive decay products can attach to dust particles that accumulate in the lungs, leading to a continuous exposure of the lung tissues to high LET alpha particles. Due to this radiation exposure, the EPA claims that radon is the second leading cause of lung cancer in the United States. Another important source of human exposure to IR is medical x-ray devices, and there is a growing concern about the dramatically increased use of whole body CT scans for diagnostic purposes. For a typical CT scan, a patient will receive about 100-fold more radiation than from a typical mammogram.24 It is recommended that the use of whole body CT scans for children be very restricted due to the elevated risk of developing radiation-induced cancer for this age group.
Cancer patients who receive radiation therapy are at risk of developing secondary tumors induced by the radiation therapy treatment.1 This is particularly a concern for young patients since (1) children are more prone to radiation-induced cancer,
(2) children have a relatively good chance of surviving the primary cancer and would have long life expectancies so a secondary tumor would have plenty of time to develop, and (3) many childhood cancers are promoted by genetic defects in DNA damage response pathways, making these patients highly prone to the genotoxic effects of radiation and subsequent secondary cancers. The most sensitive tissues for the development of secondary cancer have been found to be bone marrow (leukemia), the thyroid, breast, and lung.1
(2) children have a relatively good chance of surviving the primary cancer and would have long life expectancies so a secondary tumor would have plenty of time to develop, and (3) many childhood cancers are promoted by genetic defects in DNA damage response pathways, making these patients highly prone to the genotoxic effects of radiation and subsequent secondary cancers. The most sensitive tissues for the development of secondary cancer have been found to be bone marrow (leukemia), the thyroid, breast, and lung.1
ULTRAVIOLET LIGHT
Depending on the wavelength, UV light is categorized into UVA (320 to 400 nm), UVB (290 to 320 nm), and UVC (240 to 290 nm) radiation. Most of the UVC light emitted from the sun is absorbed by the ozone layer in the atmosphere, and, thus, living organisms are mostly exposed to UVA and UVB irradiation.
Mechanisms of Damage Induction
UVC light is more damaging to DNA than UVA and UVB because the absorption maximum of DNA is around 260 nm. UVB and UVC induce predominantly pyrimidine dimers and 6-4 photoproducts, which consist of covalent ring structures that link two adjacent pyrimidines on the same DNA strand.5 The formation of these lesions results in the bending of the DNA helix, resulting in the interference with both DNA and RNA synthesis. UVA light does not induce pyrimidine dimers or 6-4 photoproducts but can induce ROS, which in turn can form SSBs and base lesions in DNA of exposed cells.
Cellular Responses
Related posts:



Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


