INTRODUCTION
The field of myeloproliferative disorders (MPDs) has evolved considerably since the sentinel observations made by William Dameshek in 1951. He had commented in an editorial in the journal Blood: “To put together such apparently dissimilar diseases as chronic granulocytic leukemia, polycythemia, myeloid metaplasia and di Guglielmo’s syndrome may conceivably be without foundation, but for the moment at least, this may prove useful and even productive. What more can one ask of a theory?” (1).
The central feature among the MPDs is effective clonal myeloproliferation without dysplasia. Other features shared by most MPDs include involvement of a multipotent hematopoietic progenitor cell, marrow hypercellularity, predisposition to thrombosis, hemorrhage, and marrow fibrosis, and more recently, mutations in different tyrosine kinases (TKs), for example, JAK2 (Janus kinase 2), platelet-derived growth factor receptor (PDGFR), and KIT (2,3,4,5). When the concept of MPDs was first proposed, it consisted of five disorders: chronic myelogenous leukemia (CML), polycythemia vera (PV), essential thrombocythemia (ET), chronic idiopathic myelofibrosis (CIMF), and erythroleukemia. Over the years, erythroleukemia was reclassified under acute myeloid leukemia (AML). The remaining four (CML, PV, ET, CIMF) are recognized as classic MPDs.
The World Health Organization (WHO) 2001 classification assigned the classic MPDs to a broader category of chronic MPDs that also included atypical MPDs, namely, chronic neutrophilic leukemia (CNL), chronic eosinophilic leukemia/hypereosinophilic syndrome (CEL/HES), and chronic MPD, unclassifiable (MPD-U). The MPDs were, in turn, classified among one of the five categories of myeloid neoplasms, the others being: (1) AML, (2) myelodysplastic syndromes (MDS), (3) MDS/MPD, and (4) mast cell disease (MCD).
In the revised 2008 WHO classification system for chronic myeloid neoplasms, the phrase disease in both MPD and MDS/MPD has been replaced by neoplasm, reflecting the neoplastic nature of these conditions, so that MPD is now referred to as myeloproliferative neoplasm (MPN) (6). In addition, MCD is now included within the MPN category (Table 6-1). Also, CIMF has recently been renamed as primary myelofibrosis (PMF). Chronic myelogenous leukemia, characterized by the reciprocal translocation of chromosomes 9 and 22, is discussed elsewhere. Here, we discuss classic MPNs, as well as CEL/HES, CNL, and MCD, for which important advances have been made, both in the understanding the disease pathology and in clinical management.
| 1. | Acute myeloid leukemia |
| 2. | Myelodysplastic syndromes (MDS) |
| 3. | Myeloproliferative neoplasms (MPNs) |
| 3.1 Chronic myelogenous leukemia | |
| 3.2 Polycythemia vera | |
| 3.3 Essential thrombocythemia | |
| 3.4 Primary myelofibrosis | |
| 3.5 Chronic neutrophilic leukemia | |
| 3.6 Chronic eosinophilic leukemia, not otherwise categorized | |
| 3.7 Hypereosinophilic syndrome | |
| 3.8 Mast cell disease | |
| 3.9 MPNs, unclassifiable | |
| 4. | MDS/MPN |
| 4.1 Chronic myelomonocytic leukemia | |
| 4.2 Juvenile myelomonocytic leukemia | |
| 4.3 Atypical chronic myeloid leukemia | |
| 4.4 MDS/MPN, unclassifiable | |
| 5. | Myeloid neoplasms associated with eosinophilia and abnormalities of PDGFRA, PDGFRB, or FGFR1 |
| 5.1 Myeloid neoplasms associated with PDGFRA rearrangement | |
| 5.2 Myeloid neoplasms associated with PDGFRB rearrangement | |
| 5.3 Myeloid neoplasms associated with FGFR1 rearrangement (8p11 myeloproliferative syndrome) |
POLYCYTHEMIA VERA
Polycythemia vera is a clonal disorder characterized by an accumulation of phenotypically normal red cells, granulocytes, and platelets. The word polycythemia is composed of the Greek words poly (“many”), cyt (“cells”), and hemia (“blood”), indicating too many blood cells (red, white, and platelets). The term vera is from the Latin word meaning true, making a distinction between PV and a host of other conditions that can result in an increase in the number of red blood cells. The main feature of the disease is elevated red cell mass (RCM) associated with predisposition to thrombosis. Polycythemia vera occurs mainly in older adults. In a large observational study of 1,638 patients with PV, median age at diagnosis was 62.1 years, and only 4% of patients were younger than 40 years (7). The median survival is long, approximately 20 years (although inferior to the general population).
The JAK2–signal transducers and activators of transcription (JAK2-STAT) pathway has been known to play an important role in hematopoiesis, which is mediated in part by erythropoietin (Epo) and thrombopoietin (Tpo) via their cognate receptors. In 2005, four different groups identified an activating mutation in the JAK2 gene in up to 97% of patients with PV (2,3,4,5). The JAK proteins are bound to the cytoplasmic domains of type I cytokine receptors (eg, the Epo and Tpo receptors), and the binding of cytokines or growth factors (eg, Epo and Tpo) to the receptors induces dimerization and phosphorylation of the JAKs. The activated JAK then phosphorylates the cytoplasmic domains of the cytokine receptors. STATs bind to these phosphorylated receptor sites and in turn are phosphorylated by the JAKs. These phosphorylated and activated STAT molecules regulate the transcription of the target genes in the nucleus. JAK2 has two domains: JH1 (the active kinase domain) and JH2 (pseudokinase domain: inhibits kinase activity of JAK2). The most common mutation in PV is a guanine-to-thymine substitution in exon 14, resulting in a valine-to-phenylalanine substitution at position 617 (JAK2V617F). This gain of function mutation, which renders JAK2 constitutively active, results in Epo-independent proliferation of erythroid precursors. The pathologic nature of this mutation has been illustrated by many studies. Mice that receive bone marrow (BM) cells expressing the JAK2 mutation develop erythrocytosis (3). In contrast to wild-type JAK2, mutated JAK2 allows for the Epo-independent growth of cell lines in culture (3,4,5). The JAK2V617F mutation is present in approximately 95% to 97% of patients with PV and is not present in secondary polycythemia. Mutations in exon 12 of JAK2 have been identified in the remaining patients with PV who are negative for JAK2V617F mutation (8). Therefore, with current sensitive testing, almost all patients with PV should have mutations in either exon 14 or exon 12 of JAK2.
Presenting constitutional symptoms (seen in 30%-50% of patients) include headache, weakness, pruritus, fatigue, dizziness, and sweating. Thrombosis and hemorrhage due to increased blood viscosity and reduced blood flow are the most common serious complications. Mild splenomegaly is seen in up to 70% of patients. Mild leukocytosis can occur with PV, as can thrombocytosis. Thrombocytosis can lead to ocular migraine and erythromelalgia (burning pain in feet or hands associated with warmth and erythema). Some patients are asymptomatic and are diagnosed after abnormal findings on a routine blood examination. The BM is typically hypercellular with megakaryocyte pleomorphism. In the polycythemia vera study group (PVSG01) study, cellularity of the pretreatment BMs (n = 281) ranged from 36% to 100% (mean 82%), with absence of stainable iron in 94% of the patients (9). Cytogenetic abnormalities are infrequent. In two large retrospective studies of patients with PV (n = 137 and n = 133), cytogenetic studies were abnormal in 11% to 14% (trisomy 8 being the most common) and had no impact on either thrombosis risk or survival (10,11).
Thrombosis is the most serious complication of PV (Table 6-2) and is a presenting manifestation in 15% to 20% of patients (9,12). In a large study of 1,213 patients with PV, thrombosis (both arterial and venous) occurred in 41% of patients overall (64% of thrombotic events were at presentation or before diagnosis and 36% during follow-up) (12). In a more recent study of 1,545 patients diagnosed using 2008 WHO diagnostic criteria, thrombosis occurred in 23% of patients before diagnosis and in 21% during follow-up (13,14). Arterial thrombosis is more common overall than venous thrombosis. Ischemic stroke and transient ischemic attacks account for a majority of arterial thromboses at diagnosis (12,13). The overall rate of thrombotic events was estimated to occur in 2.6% to 4.4% of patients per year (12,13,15). The rate of thrombotic events increases with age (1.8/100 patients per year for those in the <40 year age group to 5.1/100 patients per year for those >70 years) (12). Older age and a previous history of thrombosis have been established as risk factors for thrombosis (12,13). In the PVSG studies, one-third of the individuals who survived the initial thrombotic event had recurrent thrombosis (16). Budd-Chiari syndrome (BCS) can be a presenting manifestation of PV. PV is the underlying cause in 50% of patients with BCS (17), and JAK2 mutation has been found in 40% to 58% of patients with BCS (18).
| At Diagnosis | At Follow-Up | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Study | No. of Patients | Asymptomatic | Major Thrombosis (%) (Arterial %, Venous %) | Bleeding (%) | Major Thrombosis (%) (Arterial %, Venous %) | Bleeding (%) | Major Thrombosis Rate | Deaths From Thrombosis (%) | Deaths From Bleeding (%) |
| PVSG01 | 431 | NR | 13.9 (61, 39) | 14.9 | 27.6 (NR, NR) | 2.7 | 31 | 5 | |
| Groupo Italiano Studio Policitemia (GISP) | 1,213 | NR | 34 (67, 33) | NR | 19 (63, 37) | NR | 3.4%/y | 29.7 | 2.6 |
| ECLAP | 1,638 | NR | 36 (75, 25) | 8.1 | 10.3 (70, 30) | 7.1 | 4.4%/y | 26 | 3.7 |
| Passamonti (2000a) | 163 | 37 | 34 (64, 36) | 3 | 18 (80, 20) | NR | 19 | 6 | |
| CYTO-PV | 365 | NR | 25 (60, 40) | 4.9 | 7.4 (56, 44) | 1.9 | 2.7%/y | 44 | NR |
| IWG-MRT | 1,545 | NR | 23 (68, 32) | 4.2 | 21 (57, 43) | 4.2 | 2.6%/y | 21 | 1.4 |
The development of myelofibrosis (MF; called post-PV MF) and AML are the two major late complications of PV. Post-PV MF develops in 10% to 20% of patients with PV and is characterized by clinical features similar to PMF (anemia, cytopenias, leukoerythroblastosis, and progressive splenomegaly). Trisomy 1q is the most common chromosomal abnormality in post-PV MF. A longer disease duration (>10 years) is associated with a 15-fold higher risk of transformation to MF (P < .0001) (15). Passamonti et al reported outcomes on a series of 647 patients with PV; 68 patients developed MF after a median of 13 years (18). The median survival for post-PV MF was 5.7 years (19). In the Efficacy and Safety of Low-Dose Aspirin and Polycythemia Vera (ECLAP) study, 22 of the 1,638 patients (1.3%) developed AML after a median of 8.4 years from the diagnosis of PV. Older age and exposure to chemotherapy (32P, busulfan, and pipobroman; P = .002), but not hydroxyurea (HU) alone, was associated with an increased risk of AML (7). In a prospective study of 338 patients, 8 developed MF and 10 developed AML after a median of 3.2 years. JAK2V617F allele burden was significantly related to the risk of developing MF but not AML (20).
The most common fatal complication in PV is thrombosis, accounting for 19% to 31% of deaths during follow-up (see Table 6-2). In a study of 1,213 patients, the most frequent fatal complications were thrombosis (30%) and cancer (15% AML, 15% other cancers) (12). In the ECLAP study (n = 1,638), the most common causes of death were cardiovascular diseases, AML, and solid tumors in 45%, 13%, and 19.5%, respectively (15). In the International Working Group for Myelofibrosis Research and Treatment (IWG-MRT) IWG-MRT study (n = 1,545), the most common causes of death were cancer (22% AML, 22% other cancers) and thrombotic complications (19.5%). As is the case with thrombotic risk, older age and history of thrombosis were associated with increased mortality (15). Leukocytosis (white blood cell [WBC] count >15 × 109/L) at diagnosis of PV has been correlated with an increased risk of thrombosis (especially myocardial infarction) (21), leukemic transformation (22), development of post-PV MF (19), and worse survival (22). In a prospective study of 338 patients with PV, only age greater than 60 had a significant effect on thrombosis risk or survival; neither leukocytosis (WBC count >11 × 109/L) nor the JAK2V617F allele burden correlated with the risk of thrombosis (20), but JAK2V617F allele burden was associated with transformation to MF. A study from the IWG-MRT suggested a new prognostic model using data from 1,545 patients with PV (14). The final model included age (57-66 or ≥67), leukocytosis (≥15 × 109/L), or venous thrombosis as independent predictors of worse survival. Independent risk factors for shorter leukemia-free survival included age greater than 61 years, abnormal karyotype, and leukocyte count ≥15 × 109/L. Previous arterial thrombosis and hypertension were associated with an increased risk of arterial thrombosis, while previous venous thrombosis and age of 65 or older were risk factors for venous thrombosis (13).
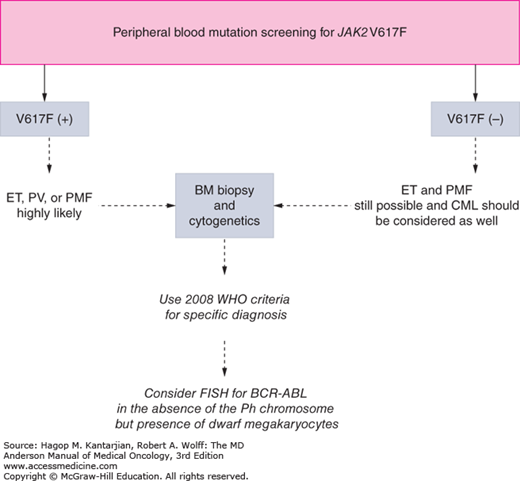
As our understanding of PV has improved with development of newer molecular markers such as JAK2 mutation, so have the diagnostic criteria for PV (Table 6-3). As JAK2V617F or similar activating mutations, such as exon 12 mutations, are present in almost 100% of patients with PV, the 2008 WHO classification appropriately incorporated JAK2 mutation as a major criterion for the diagnosis of PV. Compared to the PVSG criteria, for which RCM measurement was mandatory, the WHO criteria have placed less reliance on direct RCM measurement and established hemoglobin (Hb) cutoffs (Hb >18.5 g/dL in men or Hb >16.5 g/dL in women; Hb >17 g/dL in men and >15 g/dL in women if associated with a documented and sustained increase of at least 2 g/dL from an individual’s baseline value that cannot be attributed to correction of iron deficiency) for diagnostic purposes. This view is not universally held, and some experts still advocate use of direct RCM measurement (23). For patients suspected to have PV (based on elevated Hb/hematocrit [Hct], presence of symptoms or thrombotic/hemorrhagic complications), initial evaluation should include an analysis of peripheral blood for the JAK2 mutation and measurement of serum EPO (Fig. 6-1). As red cell proliferation is autonomous in PV, serum Epo is generally low, and erythroid colonies can grow in vitro without the addition of exogenous Epo. For patients who have not received prior chemotherapy, the endogenous (Epo-independent) erythroid colony formation test has sensitivity and specificity approaching 100%; however, the test is not commercially available. Classical morphologic features seen in BM biopsies in PV and post-PV MF are shown in Figs. 6-2,6-3,6-4.
| Major criteria |
| • Hb >18.5 g/dL in men or Hb >16.5 g/dL in women or other evidence of increased red cell volume |
| • Presence of JAK2V617F or other functionally similar mutations, such as JAK2 exon 12 mutation |
| Minor criteria |
| • Bone marrow biopsy showing hypercellularity for age with panmyelosis with prominent erythroid, granulocytic, and megakaryocytic proliferation |
| • Serum erythropoietin level below the reference range for normal |
| • Endogenous erythroid colony formation in vitro |
| Diagnosis: Both major criteria with one minor or first major with any two minor criteria |

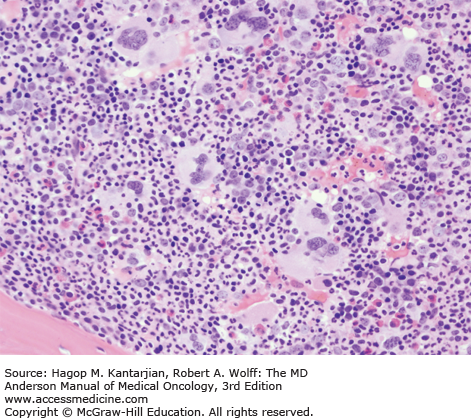
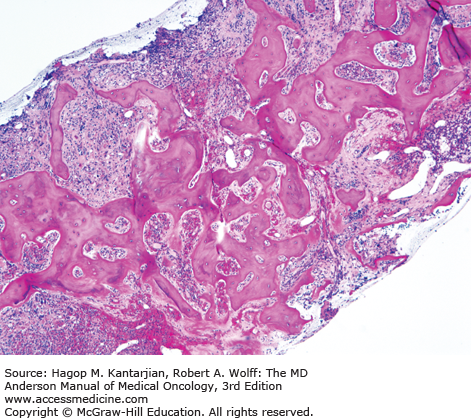
FIGURE 6-4
In contrast to the relatively normal megakaryocytes seen during early stages of PV, megakaryocytes become markedly atypical during postpolycythemic myelofibrosis phase. The atypical morphologic features include pronounced size variations, usually because of the presence of numerous small forms. Classically, megakaryocytes nuclei become hyperchromatic during this advanced stage of PV (×200).
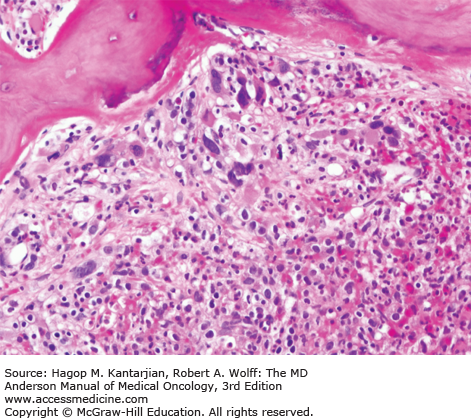
TREATMENT
The main goal of therapy is to prevent thrombotic events. The cornerstone of therapy is phlebotomy. This was established by the PVSG01 trial, in which patients were randomized to phlebotomy alone, phlebotomy plus chlorambucil, or phlebotomy plus 32P. The incidence of thrombosis during the first 2 years of the trial was significantly higher in the phlebotomy arm (23%) than in the 32P arm (16%). However, median survival was significantly higher in the phlebotomy only arm (12.6 years vs 10.9 years in the 32P arm and 9.1 in the chlorambucil arm). In addition, the AML risk was 1.5%, 9.6%, and 13.2% in the phlebotomy only, 32P, and chlorambucil arms, respectively. The incidence of MF was similar in all three arms. Given the increased risk of AML and reduced survival in the chlorambucil arm, further use of chlorambucil in PV was abandoned. The desired goal of phlebotomy is to reduce the Hct to 45% or less for males and 42% or less for females. Regular phlebotomy also induces iron deficiency, which has not been shown to be detrimental in the absence of anemia. Patients with PV who become iron deficient due to phlebotomy use should not receive iron supplementation.
The use of high-dose aspirin (acetylsalicylic acid, ASA) (900 mg daily), initially studied by PVSG, was found to increase the risk of gastrointestinal bleeding and was not pursued further (24). Low-dose ASA was proposed as an alternative after it was discovered that thrombosis in PV is mediated in part by increased thromboxane synthesis (25), and that low-dose ASA can effectively suppress its production. In the ECLAP study, Landolfi et al randomized 518 patients with PV to low-dose ASA (100 mg daily) or placebo (26). All patients had been previously treated with phlebotomy, cytoreductive therapy, or both. The use of ASA resulted in a 60% reduction in the risk of nonfatal myocardial infarction, nonfatal stroke, pulmonary embolism, major venous thrombosis, and death from cardiovascular causes (P = .03). Overall mortality, cardiovascular mortality, major venous thrombosis, and pulmonary embolism were not statistically different between the two groups. Major cerebrovascular events were less frequent in the low-dose ASA group, but the difference was not significant (3.8 vs 1.2%; P = .08). The incidence of major bleeding episodes was not significantly increased in the low-dose ASA group. A subgroup analysis indicated that ASA was more effective in patients with a disease duration of 5 years or less, platelet count less than 334 × 109/L, and Hct of 48% or greater and those who had not been treated with cytoreductive therapy. The median Hct achieved during the study was 46%, higher than the recommended targets for PV, leading to the argument that effective Hct control may lessen the beneficial effects of ASA. A recent meta-analysis from the Cochrane Hematological Malignancies Group in 630 patients with PV randomized to low-dose ASA versus placebo found nonsignificant lowering of fatal thrombotic events (odds ratio [OR] 0.20, 95% confidence interval [CI], 0.03-1.14), without excess major bleeding (27). They predicted that 19 fatal thrombotic events will be prevented for every 1,000 people treated with ASA. Therefore, all patients with PV should receive low-dose ASA unless contraindicated.
Target Hct values (<45% for men, <42% for women) for patients treated with phlebotomy or cytoreductive therapy were based on a retrospective study showing that thrombotic events increased at Hct levels greater than 44% (28). The CYTO-PV trial then evaluated the benefit of maintaining stringent control of Hct (target Hct <45%). The study randomized 365 patients with PV treated with phlebotomy or HU into two arms: one with a target Hct less than 45% and another with a target Hct between 45% and 50% (29). Patients with tight control of Hct (<45%) had a significantly lower rate of major thrombosis and death from cardiovascular causes. Hydroxyurea is the preferred cytoreductive therapy for PV patients; however, resistance or intolerance can develop in up to 13% of patients (30). Aggressive chemotherapy is not recommended. In a large study with 1,213 patients, the risk of death due to cancer was four times higher in patients who had received 32P or myelosuppressive (alkylating or nonalkylating) agents compared with those receiving phlebotomy or other pharmacological treatments (6.7% vs 1.6%; P = .06), supporting PVSG01 data (12). Similarly, in a study of 1,545 patients, exposure to pipobroman or P32/chlorambucil was associated with leukemic transformation, while exposure to HU or busulfan was not (14). In a population-based study from Sweden, exposure to two or more cytoreductive treatments (commonly HU followed by alkylating agents) was associated with a 2.9-fold increased risk of transformation to AML. However, exposure to HU alone, even at the highest dose levels, was not significantly associated with an increased risk of AML (31).
Interferon alfa (IFN-α), has been reported to be effective at suppressing erythrocytosis in 82% of patients, with a similar number reporting reduction in spleen size and alleviation of pruritus (32). Interferon alfa is not teratogenic, making it the cytoreductive therapy of choice in pregnancy. It is also nonleukemogenic (7). However, up to one-third of patients discontinue treatment due to side effects (fever, malaise, and depression).
Longer-acting pegylated forms of IFN-α (PEG-IFN-α-2a) have also been studied. Kiladjian et al reported the results of a phase II multicenter French study of PEG-IFN-α-2a in 40 patients (33). At 12 months, all 37 evaluable patients had a hematologic response, including 94.6% complete hematologic responses (CHRs), the study end point. Sequential samples tested for JAK2V617F allele burden in 29 patients showed a decrease in 26 (90%). Complete molecular response (CMR; undetectable JAK2V617F) was achieved in seven (24%) patients. Similar results were reported from another phase II study of 40 patients treated with PEG-IFN-α-2a (34). The overall hematologic response was 80%; the CHR was 70%. Of 35 patients evaluable for JAK2V617F allele burden, 54% showed a reduction and 14% achieved a CMR. After a median time on treatment of 42 months, 76% of patients had achieved a CHR and 18% a CMR (35). A newer, longer-acting form of pegylated IFN-α (PEG-proline-IFN-α-2b), which allows for subcutaneous dosing every 2 weeks, is being tested in Europe in a randomized phase III trial against HU.
The oral JAK1/2 inhibitor ruxolitinib was approved by the Food and Drug Administration (FDA) for the treatment of myelofibrosis in 2011 and for PV in December 2014, based on the results of a randomized phase III clinical trial. In the phase II trial, 34 patients refractory to HU were treated with ruxolitinib; 97% had rapid and durable normalization of Hct (<45%) by week 24. After a median follow-up of 35 months, 59% had achieved a CR (36). Responses were defined by European Leukemia Network criteria established in 2009: A CR was defined as normalization of Hct (<45% males, <42% females), leukocyte counts, platelets, and spleen size in the absence of phlebotomies and thrombotic events. There was rapid and sustained normalization of WBC and platelet counts, reduction in spleen size, and improvement in systemic symptoms (pruritus, bone pain, night sweats). Five patients experienced grade 3 or higher anemia or thrombocytopenia. The pivotal phase III RESPONSE trial randomized patients refractory to, or intolerant of, HU requiring therapy to receive either ruxolitinib 10 mg twice daily (n = 110) or best-available therapy (BAT; n = 112) (37). Twenty-one percent of patients in the ruxolitinib arm achieved the primary end point (Hct control without phlebotomy and a ≥35% reduction in spleen volume from baseline by magnetic resonance imaging at week 32) versus 1% in the HU arm. Overall, 77% of patients in the ruxolitinib group achieved one or more of the criteria for the primary end point, and 49% (vs 1% of BAT patients) had a 50% or greater improvement in the MPN-Symptom Assessment Form total symptom score, a validated measure of patient-reported symptoms (38). Ninety-six patients crossed over to the ruxolitinib arm after week 32. Grade 3/4 anemia or thrombocytopenia occurred in 1.8% and 5.5% of patients, respectively, compared with 0% and 3.6% of BAT patients, respectively. Ruxolitinib has been approved as therapy for PV after resistance to or intolerance of HU.
All patients with PV should undergo phlebotomy and receive low-dose ASA unless contradicted. Patients who are at high risk for thrombosis (age >60 years or history of thrombosis) should receive cytoreductive therapy (HU preferred; IFN-α can be considered, especially PEG-IFN-α-2a, in younger patients). The goal Hct is 45% or less for males and 42% or less for females. With the approval of ruxolitinib for PV, patients resistant or intolerant to HU should be offered ruxolitinib, a JAK2 inhibitor.
ESSENTIAL THROMBOCYTHEMIA
Essential thrombocythemia is characterized by persistent thrombocytosis with a predisposition to thrombosis and bleeding. Essential thrombocythemia is not a cytogenetically or a morphologically defined disease entity and remains a diagnosis of exclusion. It is a disease of the elderly; the median age of diagnosis is 55 to 60 years. The female-to-male ratio is 2:1.
Reactive causes of thrombocytosis should be excluded. Most often, the underlying cause is apparent (postsplenectomy, acute infection, blood loss). Other MPNs, such as PV or CML, can also present with thrombocytosis and should be ruled out. In a population-based study (ages 18 to 65 years), 99 of the 9,998 persons studied (1%) had a platelet count greater than 400 × 109/L at baseline, of whom only 8 (0.1% of the population studied) had persistent thrombocytosis at greater than 6 months (39). Three of the eight patients were confirmed to have ET at baseline, with one additional case of ET diagnosed after 5 years of follow-up. In another study of 732 patients with thrombocytosis (>500 × 109/L), ET was present in 5.5% and reactive thrombocytosis in 87% patients (40). The magnitude of elevation in the platelet count does not distinguish between reactive and clonal thrombocytosis. In 280 consecutive patients with extreme thrombocytosis (platelet count >1 million), reactive thrombocytosis was noted in 82%, and 14% had MPNs, including ET in 4% (41). Reactive thrombocytosis, irrespective of the degree of elevation of platelet counts, does not increase the risk of thromboembolic or bleeding complications. Such complications, if seen, are the results of underlying disease conditions (malignancy, iron deficiency from gastrointestinal bleeding) rather than of elevated platelets.
Thrombopoietin regulates the differentiation and proliferation of megakaryocytes. It is produced primarily by the liver parenchymal cells. The gene for Tpo is located on chromosome 3q27-28. It binds to the c-Mpl receptors on platelets and megakaryocytes. When the platelet count is low, more free Tpo is available to bind to megakaryocytes to stimulate proliferation, leading to a rise in the platelet count and vice versa. In most cases of reactive thrombocytosis, Tpo is increased via acute phase reactants, such as interleukin 6. Unlike PV, for which Epo levels are generally low, Tpo levels are high normal or abnormally increased in ET (42). This may be due to the increased BM stromal production of Tpo or decreased clearance, as expression of platelet c-Mpl is markedly reduced in ET (42). Approximately 50% of patients with ET harbor the JAK2V617F mutation, and 3% to 5% have a mutation in the thrombopoietin receptor (MPL) (W515L/K; tryptophan to leucine or lysine substitution at residue 515) (43), both of which lead to dysregulated JAK-STAT signaling. Most patients with ET have JAK2V617F allele burdens less than 50%, suggesting that lower JAK2V617F gene dosage may lead to the development of ET versus PV, for which the allele burden is generally higher (44).
Recently, mutations in the gene encoding calreticulin (CALR) have been found in nearly 70% of patients with ET negative for JAK2 or MPL mutations (~25% of all patients with ET) (45,46). The CALR mutation may define a distinct subset of ET. In a series of 717 patients with ET, those with CALR mutations were younger and predominantly male and had a lower incidence of thrombosis, lower hemoglobin levels, lower leukocyte counts, and higher platelet counts than those with the JAK2V617F mutation (47). Additional studies are needed to fully elucidate the implications of molecular studies in clinical practice.
With the increasing use of automated blood counters and routine blood count screenings, more patients with ET are being diagnosed while asymptomatic. Constitutional symptoms are uncommon in ET. Vasomotor manifestations such as dizziness, lightheadedness, acral paresthesia, livedo reticularis, and erythromelalgia were noted in 34% of patients in one study of 147 patients with ET (48). Mild splenomegaly (<5 cm) was noted in up to 40% of patients, leukocytosis in 30% to 40%, and mild anemia in 10% to 20%. Thromboembolic and bleeding complications are the major cause of morbidity and mortality in ET. A series of 322 patients with ET from one institution reported a 26% incidence of major thrombosis and 11% incidence of major bleeding at diagnosis (49). Hemorrhagic complications increase with extreme thrombocytosis (platelet count >1.5 million/μL) and with the use of antiplatelet therapy such as ASA.
Most serious late complications of ET include transformation to AML and MF (post-ET MF). In a study of 605 patients with ET, the incidence of AML transformation was 3.3%, with a median time to transformation of 11.5 years (50). Risk factors for transformation included anemia, platelet count greater than 1,000 × 109/L, and increasing age. JAK2 mutational status or the type of therapy (including HU) did not influence the risk of leukemic transformation. In another series of 195 patients with ET, the median time to transformation to MF was 8 years, with an actuarial probability of 2.7% at 5 years, 8.3% at 10 years, and 15.3% at 15 years (51). In a large series from seven centers in Europe, patients were reclassified using 2008 WHO criteria (52). The diagnosis of ET was confirmed in 891 patients and revised to early/prefibrotic MF in 180 patients. Among patients with ET, the incidence of transformation to AML and MF was 1% and 4%, respectively. The 15-year cumulative incidence of transformation to AML or MF was 2.1% and 9.3%, respectively. In another series of 576 patients with ET, the cumulative incidence of AML was 3.8% and of transformation to MF was 9.5% (53).
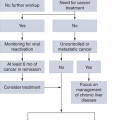
Essential thrombocythemia remains a diagnosis of exclusion. Reactive thrombocytosis must be excluded. An important change in the 2008 WHO classification was lowering the platelet count for ET diagnosis from 600 × 109/L to 450 × 109/L (Table 6-4). Bone marrow biopsy is mandatory for diagnosis to rule out PMF and MDS/MPN. A BM biopsy typically shows large but mature-appearing megakaryocytes with deeply lobulated or hyperlobulated nuclei (Fig. 6-5). The peripheral smear is significant for markedly increased platelets (Fig. 6-6). Reticulin staining should be done to rule out any underlying fibrosis. The presence of megakaryocytic dysplasia in the BM biopsy suggests “prefibrotic” MF, which implies a higher risk of transformation to overt MF or AML (52). Chronic neutrophilic leukemia should be ruled out by testing for the Bcr-Abl fusion gene. Testing for the JAK2, MPL, or CALR mutations is recommended to establish the clonal nature of the disease, ruling out reactive thrombocytosis (Fig. 6-7).
| 1. Sustained platelet count ≥450 × 109/L |
| 2. Bone marrow biopsy specimen showing proliferation mainly of the megakaryocytic lineage with increased numbers of enlarged, mature megakaryocytes; no significant increase or left shift of neutrophil granulopoiesis or erythropoiesis |
| 3. Not meeting WHO criteria for PV, PMF, CML, MDS, or other myeloid neoplasm |
| 4. Demonstration of JAK2V617F or other clonal marker, or in the absence of a clonal marker, no evidence for reactive thrombocytosis |
| Diagnosis of ET requires meeting all four criteria |
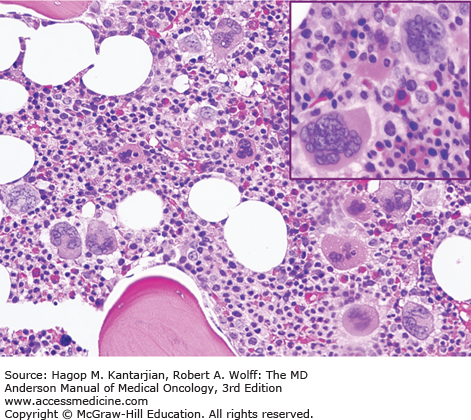

FIGURE 6-7
Diagnostic algorithm for suspected essential thrombocytosis. FISH, fluorescence in situ hybridization. (Reproduced with permission from Tefferi A, Vardiman JW. Classification and diagnosis of myeloproliferative neoplasms: the 2008 World Health Organization criteria and point-of-care diagnostic algorithms. Leukemia. 2008;22:14-22.)
Like in PV, thrombosis and hemorrhage are the main complications of ET. Older age and history of prior thrombosis have been shown to predict for future thrombotic events in most studies, whereas cardiovascular risk factors have been predictive in only some. Platelet count does not correlate with risk of thrombosis in ET (Table 6-5). Some studies have found an inverse relationship between the platelet count and the thrombotic risk. This is thought to be due to acquired von Willebrand factor (vWF) disease with elevated platelet counts (eg, >1.5 million/μL), predisposing to more bleeding and protection from thrombosis.
| Risk Factors Studied | |||||||
|---|---|---|---|---|---|---|---|
| Study | No. of Patients | Age >60 Years, Odds Ratio/Hazard Ratio/Significance Level | History of Thrombosis | Risk Factors for CV Events (Smoking, Diabetes, Hypertension, Hyperlipidemia) | Platelet Count >1,000 × 109/L | Leukocytosis | JAK-2 Status |
| Colombi (1991) (60a) | 103 | NS | P < .001 | – | NS | – | – |
| Cortelazzo (1990) (60b) | 100 | 10.3 | 13 | NS | NS | – | – |
| Besses (1999) (60c) | 148 | 3.3 | 3.0 | 4.7 | NS | – | – |
| Bazzan (1999) (60d) | 187 | NS | – | NS | NS | – | – |
| Jantunen (2001) (60e) | 132 | NS | – | P = .01 | NS | – | – |
| Chim (2005) (60f) | 231 | P = .01 | NS | – | NS | – | – |
| Wolanskyj (49) | 322 | 1.51 | 2.3 (arterial only) | NS | – | 1.74 (WBC > 15,000) | NS |
| Carobbio (2007) (60g) | 439 | 2.3 (age and previous thrombosis evaluated together) | 2.3 | – | NS | 2.3 (WBC > 8,700) | NS |
| Alvarez- Larran (2007) (60h) | 126 (<40 y) | NA | NS | Smoking | NS | – | NS |
| Radaelli (2007) (60i) | 306 | NS | 7.6 | P < .05 | NS | – | – |
| Tefferi (2007) (60j) | 605 | NS | P < .001 | NS | – | WBC (≥15,000) P < .01 for thrombosis at baseline (NS for thrombosis on follow-up) | NS |
| Passamonti (2008) (60k) | 605 | P < .001 | P = .03 | NS | NS | NS | – |
| Carobbio (56) | 1,063 | 1.7 (age and previous thrombosis evaluated together) | 1.7 | – | Patients with WBC <11,000 and platelet <1,000: most likely to have JAK-2 mutation and highest risk of thrombosis | ||
| Carobbio (55) | 891 | 1.5 | 1.93 | 1.56 | 0.50 | 1.14 | 2.04 |
Recent studies from the IWG-MRT of a series of 891 patients, from seven centers in Europe diagnosed with ET using 2008 WHO criteria, reported a 6% incidence of major bleeding at a rate of 0.79 patients/year (54). The incidence of thrombosis (fatal and nonfatal events) was 25% at a rate of 1.9% of patients/year (52,55). The rate of nonfatal arterial events (1.2% of patients/year) was higher than that of venous events (0.6% patient/year) (55). Factors independently associated with bleeding included previous hemorrhage and ASA therapy (54). Factors independently associated with major thrombosis included age greater than 60 years; cardiovascular risk factors (diabetes, hypertension, or smoking); previous thrombosis; and JAK2V617F mutation (55). Leukocytosis (>11 × 109/L) was an additional independent risk factor for arterial thrombosis; male gender increased the risk of venous thrombosis. Extreme thrombocytosis (platelet count > 1,000 × 109/L) was independently associated with a reduced risk of arterial thrombosis (55). This is thought to be due to acquired vWF disease with elevated platelet counts, predisposing to more bleeding and protection from thrombosis, which is consistent with previous reports showing an inverse relationship between platelet count and thrombotic risk (56).
Using these risk factors, a new prognostic model was proposed to predict risk of thrombosis in patients with ET (International Prognostic Score of Thrombosis in Essential Thrombocythemia [IPSET-Thrombosis]) (57). The prognostic score assigned weights to the four risk factors, and the patients could be stratified in three risk categories, with an annual risk of thrombosis ranging from 1.03% of patients/year for the low-risk group to 3.56% of patients/year for the high-risk group (Table 6-6).
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


