Symptoms
Frequency
Signs
Frequency
Headache
++++
Hypertension
++++
Palpitations
+++
– Sustained
++
Sweating
+++
– Paroxysmal
++
Anxiety/nervousness
++
Tachycardia or reflex bradycardia
+++
Abdominal/chest pain
++
Sweating/diaphoresis
+++
Nausea
++
Orthostatic hypotension
+++
Tremulousness
++
Pallor
++
Fatigue/weakness
++
Fever/hypermetabolism
++
Dyspnea
+
Hyperglycemia
++
Dizziness/faintness
+
Vomiting
++
Heat intolerance
+
Weight loss
++
Pain/paresthesias
+
Increased respiratory rate
++
Visual symptoms
+
Flushing
+
Constipation
+
Convulsions
+
Diarrhea
+
Psychosis (rare)
+
Patients with primarily epinephrine-secreting PHEOs/PGLs more frequently display signs and symptoms compared to those with norepinephrine-producing tumors. Patients with dopamine-secreting PHEOs/PGLs usually present with less typical symptoms, such as hypotension, diarrhea, and weight loss. However, the severity of symptoms does not necessarily correlate with plasma catecholamine levels [7]. Clinical symptoms can mimic a number of different conditions (Table 12.2) and they vary from patient to patient. Moreover, approximately 8–13% of patients may be completely asymptomatic, usually due to a small (less than 5 mm) tumor or a dedifferentiated tumor without catecholamine-synthesizing enzymes [13, 14]. As a result, PHEOs/PGLs are often missed and are not discovered until autopsy [5, 10, 15]. Patients sometimes present with potentially life-threatening conditions due to excessive catecholamine release from a tumor (Table 12.3). PHEO/PGL-induced hemodynamic or metabolic attacks are variable in duration and frequency. They can occur daily or as infrequently as once a few months, lasting from seconds to several hours.
Table 12.2
Differential diagnosis of PHEO/PGL
System | Diagnosis |
|---|---|
Endocrine | Adrenal medullary hyperplasia Hyperthyreosis, thyrotoxicosis Carcinoid Hypoglycemia, insulin reaction Medullary thyroid carcinoma Hyperadrenergic essential hypertension Mastocytosis Menopausal syndrome |
Cardiovascular | Heart failure Arrhythmias Ischemic heart disease, angina pectoris Myocardial infarction Mitral valve prolapse Abdominal catastrophe/aortic dissection Baroreflex failure Syncope Orthostatic hypotension Labile hypernoradrenergic essential hypertension Renovascular disease |
Neurological | Migraine or cluster headaches Stroke Diencephalic autonomic epilepsy Meningioma Paroxysmal tachycardias including postural tachycardia syndrome Guillain-Barré syndrome Encephalitis Intracranial lesions Cerebral vasculitis and hemorrhage |
Psychogenic | Anxiety or panic attacks Factitious use of drugs Somatization disorder Hyperventilation |
Pharmacologic | Tricyclic antidepressant Cocaine Amphetamine Alcohol withdrawal Drugs stimulating adrenergic receptors Abrupt clonidine withdrawal Dopamine antagonists Ingestion of tyramine-containing foods or proprietary cold preparations while taking monoamine oxidase inhibitors Ephedrine-containing drugs Factitious use of various drugs including catecholamines |
Other | Neuroblastoma, ganglioneuroma, ganglioneuroblastoma Acute intermittent porphyria Mastocytosis Unexplained flushing spells Recurrent idiopathic anaphylaxis Toxemia of pregnancy Unexplained shock Lead or mercury poisoning |
Table 12.3
Emergency situations associated with PHEO/PGL
Clinical setting | Symptoms |
|---|---|
Pheochromocytoma multisystem crisis (PMC) | Hyper- and/or hypotension Multiple organ failure Body temperature ≥ 40 °C Encephalopathy |
Cardiovascular | Collapse Hypertensive crisis Hypertensive crisis upon induction of anesthesia Hypertensive crisis induced by medication or other mechanisms Shock or profound hypotension Acute heart failure Myocardial infarction Arrhythmia Cardiomyopathy Myocarditis Dissecting aortic aneurysm Limb and/or organ ischemia, digital necrosis, or gangrene |
Pulmonary | Acute pulmonary edema Adult respiratory distress syndrome Pulmonary hypertension |
Abdominal | Abdominal bleeding Paralytic ileus Acute intestinal obstruction Severe enterocolitis and peritonitis Colon perforation Bowel ischemia and generalized peritonitis Mesenteric vascular occlusion Acute pancreatitis Cholecystitis Megacolon Watery diarrhea syndrome with hypokalemia |
Neurological | Hemiplegia Limb weakness General muscle weakness Generalized seizures Stroke |
Renal | Acute renal failure Acute pyelonephritis Severe hematuria Renal artery stenosis by compression of tumor |
Metabolic | Diabetic ketoacidosis Lactic acidosis |
Ocular | Acute blindness Retinopathy |
The classical PHEO/PGL symptoms include headaches, profuse sweating, and palpitations. A high number of patients suffer from sustained or paroxysmal hypertension [6, 10]. If these symptoms are present together, they are highly suggestive for PHEO/PGL [7, 10, 13, 18, 19].
Headaches are the most prevalent symptom (up to 90%) in patients with PHEO/PGL. Seriousness of headaches varies from mild to severe, and they can last up to several days [20]. Sweating and diaphoresis occurs in 60–70% of PHEO/PGL patients [7, 20]. Catecholamine effects, specifically epinephrine, on cardiac β-adrenoceptors can manifest as palpitations [7].
Sustained or paroxysmal hypertension, often resistant to treatment, is present in around 90% of PHEO/PGL patients. Those with sustained high blood pressure present disturbances in the diurnal blood pressure rhythm, reflected by the lack of nocturnal blood pressure dip [21, 22]. Hypertensive PHEO/PGL patients may also exhibit decreased cardiac output [23] or can present with an acute catecholamine cardiomyopathy/myocardial damage [24–26]. Severe hypertension may result in emergency situations requiring immediate medical attention and treatment [7]. Hypertensive crisis and symptoms associated with paroxysmal blood pressure elevations can occur due to excessive catecholamine release triggered by accidental tumor manipulation during diagnostic procedures (e.g., endoscopy), an increase in intra-abdominal pressure (e.g., palpation, defecation, urination, accident), and administration of anesthesia or certain drugs (Table 12.4) or through ingestion of food and beverages containing tyramine (certain cheeses, beers, wines, bananas, chocolate) or synephrine (citrus fruit juice) [10, 28, 30–34].
Table 12.4
Medications contraindicated in patients with known or suspected PHEO/PGL
Drug class | Examples | Relevant clinical uses |
|---|---|---|
β-Adrenergic receptor blockersa | Propranolol, sotalol, timolol, nadolol, labetalol | But may be used to treat conditions that result from catecholamine excess (hypertension, cardiomyopathy, heart failure, panic attacks, migraine, tachycardia, cardiac arrhythmias) |
Dopamine D2 receptor antagonists including antipsychotics | Metoclopramide, sulpiride, amisulpride, tiapride, chlorpromazine, prochlorperazine, droperidol | Control of nausea, vomiting, psychosis, hot flashes, tranquilizing effects |
Tricyclic antidepressants and norepinephrine reuptake inhibitors | Amitriptyline, imipramine, nortriptyline, clomipramine | Treatment of insomnia, neuropathic pain, nocturnal enuresis in children, headaches, depression (rarely) |
Other antidepressants (serotonin reuptake inhibitors) | Paroxetine, fluoxetine, duloxetine | Depression, anxiety, panic attacks, antiobesity agents |
Monoamine oxidase inhibitors | Tranylcypromine, moclobemide, phenelzine | Nonselective agents rarely used as antidepressants (owing to “cheese effect”) |
Sympathomimeticsa | Ephedrine, pseudoephedrine, fenfluramine, amfepramone, phendimetrazine, methylphenidate, phentermine, dexamfetamine | Control of low blood pressure during surgical anesthesia, as decongestants, antiobesity agents |
Chemotherapeutic agentsa | Antineoplastic actions and treatment of malignant pheochromocytoma | |
Oxazolidinone antibiotics | Linezolid | Treatment of infections caused by multiresistant gram-positive bacteria |
Opioid analgesicsa and naloxone | Morphine, pethidine, tramadol, oxycodone, heroin | Induction of surgical anesthesia |
Neuromuscular blocking agentsa | Succinylcholine, tubocurarine, atracurium | Induction of surgical anesthesia |
Peptide and steroid hormonesa | ACTH, glucagon, dexamethasone, prednisone, hydrocortisone, betamethasone | Diagnostic testing |
Illegal recreational drugs | Ketamine, cocaine | |
Chewing tobacco |
Although hypertension is the most common clinical sign, some patients (up to 10%) may have normal blood pressure or may present with hypotension, particularly postural hypotension, or alternating episodes of hyper- and hypotension [13, 35–37]. Orthostatic hypotension is usually accompanied by orthostatic tachycardia and is seen in epinephrine-secreting PHEO/PGL.
Other PHEO/PGL symptoms include flushing or pallor, nausea and vomiting (often exercise induced), anxiety or panic attacks, dyspnea, weight loss despite normal appetite, warmth with or without heat intolerance, or general weakness [6, 7, 35]. Less commonly, PHEO/PGL presents as fever of unknown origin, constipation due to catecholamine-induced decrease in intestinal motility, or cholesterol gallstones [38–40]. Due to the metabolic effects of epinephrine, hyperglycemia with low levels of plasma insulin associated with hypertensive episodes can occur. PHEOs/PGLs can also cause insulin resistance and diabetes mellitus manifestation [7, 41–47]. Rarely, PHEOs/PGLs can produce vasoactive intestinal peptide resulting in watery diarrhea, hypokalemia, and achlorhydria [48].
Patients may also complain of symptoms resulting from compression of tissues surrounding the tumor. For example, tumors located in the abdomen or chest can cause abdominal or chest pain. Patients with PGLs of the neck can present with dysphagia and dysphonia, and those with tumors growing in the head and neck area can display tinnitus, hearing loss, or cranial nerve palsy [49].
Since PHEO/PGL can have potentially life-threatening consequences, recognizing the signs and symptoms of these tumors leading to appropriate diagnostic sequence is critical. Evaluation for PHEO/PGL should be warranted in patients: (a) with a family history of PHEO/PGL or certain hereditary cancer syndromes (Table 12.5); (b) presenting with hypertension, tachycardia, sweating, and pallor; (c) presenting with resistant hypertension; (d) presenting with any paroxysmal symptoms; (e) presenting with hypertension and other symptoms in response to examination, anesthesia, surgery, certain medications, or foods and drinks; and (f) with adrenal incidentalomas [7, 11, 17, 51].
Table 12.5
Susceptibility genes and hereditary cancer syndromes associated with PHEO/PGL development and genotype-phenotype correlations
Molecular cluster | Gene | Locus | Mutation type | Inheritance | Syndrome | PHEO/PGL penetrance | Biochemical phenotype | Typical tumor localization | Malignancy rate | Associated clinical characteristics/other tumors |
|---|---|---|---|---|---|---|---|---|---|---|
Krebs cycle (cluster 1a) | SDHA | 5p15 | Germline | AD | PGL5 | Unkno wn | Unknown | HNPGL/TAPGL | 0–14% | • Homozygots: Leigh’s syndrome • Clear cell renal carcinoma • Gastrointestinal stromal tumors • Pituitary adenomas |
SDHB | 1p36.13 | Germ line and somatic | AD | PGL4 | 30–100% | MN, NMN, MTY, NS | TAPGL; rarely HNPGL/adrenal | 31–71% | • Clear cell renal carcinoma • Gastrointestinal stromal tumors • Pituitary adenomas • Possibly breast carcinoma • Possibly papillary thyroid carcinoma | |
SDHC | 1q23.3 | G ermline | AD | PGL3 | Unknown | MN, NMN, MTY, NS | HNPGL; rarely TAPGL/adrenal | Low | • Clear cell renal carcinoma • Gastrointestinal stromal tumors • Pituitary adenomas | |
SDHD | 11q23 | Germline and somatic | AD PI | PGL1 | 73–90% | MN, NMN, MTY, NS | HNPGL; rarely TAPGL/adrenal | Low (<5%) | • Clear cell renal carcinoma • Gastrointestinal stromal tumors • Pituitary adenomas | |
SDHAF2 | 11q12 | Germline | AD PI | PGL2 | 100% | Unknown | HNPGL | Unknown | ||
FH | 1q42.1 | Germline | – | Unkno wn | NMN | Adrenal/TAPGL | Unknown (High?) | • Leiomyomatosis of skin and uterus • Clear cell renal carcinoma | ||
MDH2 | 7q11.23 | Germline | – | Unknown | Unknown (NMN?) | Unknown (TAPGL?) | Unknown | |||
Pseudohypoxic (cluster 1b) | VHL | 3p25.3 | Germline a nd somatic | AD | Von Hippel-Lindau | 10–20% | NMN | Adrenal; rarely TAPGL/HNPGL | Low (<5%) | • Hemangioblastomas • Clear cell renal carcinoma • Tumors of pancreatic islets • Retinal angioma • Retinal, pancreatic, and testicular cysts |
HIF2A | 2p21 | G ermlinea and somatic | – | Pacak-Zhuang syndrome | Unknown | NMN | TAPGL/adrenal | Unknown | • Somatostatinoma, often multiple • Polycythemia • Eye changes • Organ cysts | |
PHD1/ EGLN2 | 19q13.2 | Germline | – | Unknown | NMN | Unknown | Unknown | • Polycythemia | ||
PHD2/ EGLN1 | 1q42.2 | Germline | – | Unknown | NMN | Unknown (TAPGL?) | Unknown | • Polycythemia | ||
Kinase signaling (cluster 2) | NF1 | 17q11.2 | Germline and somatic | AD | Neurofibromatosis type 1 | <6% | MN, NMN | Adrenal; rarely TAPGL | 11% | • Café au lait spots • Neurofibromas • Freckles • Benign hamartomas o f iris (Lisch nodules) • Gliomas and optical gliomas • Duodenal somatostatinomas • Sphenoid dysplasia/pseudoarthritis |
RET | 10q11.21 | G erm line and somatic | AD | MEN2 | 50% | MN, NMN | Adrenal | Low (<1–5%) | • Medullary thyroid carcinoma • Hirschsprung’s disease • Hyperparathyreosis/hypercalcemia (MEN2A) • Marfanoid habitus, ganglioneuromas (MEN2B) | |
TMEM127 | 2q11.2 | Germ line | – | Unknown | MN and NMN | Adrenal/TAPGL/HNPGL | Low (4%) | • Possibly breast carcinoma • Possibly papillary thyroid carcinoma | ||
MAX | 14q23.3 | Germline and s omatic | – | Unknown | NMN and MN | Adrenal | 10–25% | • Neuroblastoma | ||
KIF1Bβ | 1p36.22 | Germline | – | Unknown | Unknown | Unknown (adrenal?) | Unknown | • Lung, colorectal adenocarcinoma • Neuroblastoma | ||
H-RAS | 11p15.5 | S omatic | – | N/A | MN, NMN | Adrenal/TAPGL | Unknown | |||
K-RAS | Somatic | – | N/A | Unknown | Adrenal | Unknown | ||||
ATRX | Xq21.1 | Somatic | – | N/A | Unknown | Unknown (adrenal? TAPGL?) | Unknown |
Biochemical Diagnosis of Pheochromocytoma
Initial Biochemical Testing
Most PHEOs and PGLs are characterized by excessive production of catecholamines, and thus, biochemical evidence of catecholamine production is an elementary step in diagnosis. Historically, biochemical diagnosis of PHEO/PGL relied on the measurement of urinary and plasma catecholamines (epinephrine and norepinephrine) together with measurement of urinary levels of catecholamine metabolites and vanillylmandelic acid. These tests can often lead to false negatives due to fluctuating levels of catecholamine release in many PHEOs/PGLs [10, 28]. Since PHEOs/PGLs often secrete catecholamines episodically, plasma or urinary levels of catecholamines may be normal. Approximately 30% of PHEOs/PGLs do not secrete catecholamines, even if they still synthesize them or they do not secrete catecholamines in amounts sufficient enough to produce the classical clinical presentation of a tumor with positive test results [52, 53]. Moreover, catecholamines are normally produced by the adrenal medulla and sympathetic nerves. Thus, high catecholamine levels are present in multiple different diseases and conditions and are not specific for these tumors [53].
On the other hand, metanephrines, the O-methylated metabolites of catecholamines, are produced continuously within PHEO/PGL cells, and their production is independent of catecholamine release [54, 55]. Therefore, diagnostic evaluation of plasma-free or urine-fractionated (i.e., normetanephrine and metanephrine measured separately) metanephrines is preferred and is currently the most sensitive diagnostic test (97% sensitivity and 93% specificity for measurement for plasma-free metanephrines) [8, 56–63]. Plasma metanephrines are usually measured in the free form. Metanephrines in urine are measured after deconjugation, although measurement of urine-free metanephrines is also possible [64, 65].
When interpreting the results, differentiating between a mild and high increase in catecholamine or metanephrine levels is very important. In patients with biochemically active tumors, an increase is usually two to four times higher than the upper reference limit. Mildly elevated levels of catecholamines are mostly due to interfering medications [10] (Table 12.6). If it is difficult to distinguish an increased catecholamine release due to sympathetic activation or from the presence of PHEO/PGL, a clonidine suppression test can be performed [66, 67]. Under physiologic conditions, clonidine suppresses release of neuronal norepinephrine (and so normetanephrine). A decrease in elevated plasma normetanephrine levels by ≥40% or within the reference limits after clonidine is administered indicates that sympathetic activation is the cause of elevation. Failure to depress plasma normetanephrine supports the presence of PHEO/PGL [67]. The clonidine suppression test has a high diagnostic sensitivity when combined with measurement of plasma normetanephrine responses to suppression. False positive elevations in plasma normetanephrine levels can be accurately identified with the combination of these tests. However, the reliability of the test can be compromised by tricyclic antidepressants and diuretics [42], as well as in patients with normal or only mildly elevated plasma catecholamine levels, despite the presence of PHEO/PGL [8].
Table 12.6
Compounds that may cause false-positive elevations of plasma and urinary catecholamines or metanephrines
Compound group | Examples | Catecholamines | Metanephrines | ||
|---|---|---|---|---|---|
NE | E | NMN | MN | ||
Tricyclic antidepressants | Amitr iptyline, imipramine, nortriptyline | +++ | − | +++ | − |
α-Blockers (nonselective) | Phenoxybenzamine | +++ | − | +++ | − |
α-Blockers (α1-selective) | Doxaz osin, terazosin, prazosin | + | − | − | − |
β-Blockers | Atenolol, metoprolol, propranolol, labetalol | + | + | + | + |
Calcium channel antagonists | Nifedipine, amlodipine, diltiazem, verapamil | + | + | − | − |
Vasodilators | Hydralazine, isosorbide, minoxidil | + | − | ? | ? |
Monoamine oxidase inhibitors | Phenelzin, tranylcypromine, selegiline | − | − | +++ | +++ |
Sympathomimetics | Ephedrine, pseudoephedrine, amphetamines, albuterol | ++ | ++ | ++ | ++ |
Stimulants | Caffeine, nicotine, theophylline | ++ | ++ | ? | ? |
Miscellaneous | Levodopa, carbidopa Cocaine | ++ ++ | − ++ | ? ? | ? ? |
In addition to measurement of metanephrines, recent evidence suggests that plasma 3-methoxytyramine is a biomarker for dopamine-producing tumors. Although it is currently only available in certain research centers, measurements of this biomarker are valuable for detecting very rare, exclusively dopamine-producing tumors (due to the lack of dopamine β-hydroxylase), which can be easily overlooked by solely measuring metanephrines [68, 69]. Moreover, methoxytyramine can also serve as an indicator of malignancy [4]. Exclusively dopamine-secreting tumors can also be detected by measuring serum levels of dopamine. Measurement of urinary dopamine levels is not useful, since it reflects dopamine production in the renal tubules, not in a potential tumor [27].
A nonspecific biomarker of neuroendocrine tumors, chromogranin A , is often measured in PHEO/PGL patients. Chromogranin A is commonly secreted by chromaffin cells, and its levels are elevated in 91% of patients with PHEO/PGL [70]. Despite its nonspecificity, chromogranin A , in combination with catecholamine measurement, can facilitate the diagnosis of PHEO/PGL, especially in tumors related to mutations in succinate dehydrogenase (SDH) subunit B gene [71, 72]. Chromogranin A is also a helpful diagnostic tool in patients with biochemically silent tumors and in disease monitoring [73, 74].
In very rare cases, PHEOs/PGLs can co-secrete other hormones, for example, ACTH or cortisol. These patients often present with the clinical picture of Cushing disease in addition to PHEO/PGL [75–77].
Determining the biochemical phenotype of PHEO/PGL can also be helpful in navigating further investigation and treatment, specifically localization of the tumor by imaging studies, genetic screening, determining the presence of metastatic disease, and an appropriate adrenergic blockade. Based on the type of catecholamines secreted, PHEOs/PGLs can be divided into three basic biochemical phenotypes: (a) adrenergic (epinephrine/metanephrine), (b) noradrenergic (norepinephrine/normetanephrine), and (c) dopaminergic (dopamine/methoxytyramine). Mixed phenotypes are common, and in such cases, PHEO/PGL-producing metanephrine and normetanephrine are considered adrenergic, and those secreting normetanephrine and methoxytyramine are considered dopaminergic. Since epinephrine/metanephrine are produced almost exclusively (99%) in the adrenal gland, adrenergic biochemical phenotype is typical for PHEO (Table 12.5).
Follow-Up Biochemical Testing
Although PHEOs/PGLs are rare tumors, a large number of patients are tested for these tumors in the process of differential diagnosis for secondary hypertension as well as other diseases. Because of this, false-positive results are expected, and they may outnumber true-positive results, even when tests with high specificities are used. This requires follow-up biochemical testing in patients with initially positive results, to confirm or rule out PHEO/PGL. However, when judging the likelihood of a PHEO/PGL from a single test, the degree of initial clinical suspicion or pretest probability of the tumor should be taken into account, which impacts the posttest probability of a tumor [7]. Moreover, patients with known hereditary syndromes associated with PHEO/PGL or with a history of tumors should periodically undergo screening. Biochemical testing is also used to confirm the success of surgical treatment and to evaluate activity of the disease in patients with metastases.
Sample Collection and Test Interferences
To ensure the reliability of biochemical test results, it is crucial to guarantee certain conditions during blood sample collection . Before collection of blood for measurements of plasma-free metanephrines, patients should be lying supine in a quiet room for at least 20–30 min with a previously inserted intravenous line (to minimize sympathoadrenal activation associated with venipuncture or upright posture) [53, 78]. Alternatively, with a higher risk of false-positive results, the sample may be collected from a seated patient, provided that upper reference limits obtained after supine rest are used [79]. If seated test returns back positive, it should be repeated after rest in supine position to rule out false positivity of initial test [8].
Although 24-h urine collection seems to solve the problem with the rigid conditions needed for blood sampling, it is not that simple. Twenty-four-hour urine results are not always accurate because of unreliable collection from the patient. Samples are also often influenced by diet and the activation of sympathoneuronal and adrenal medullary systems (e.g., during physical activity or changes of posture). To ensure more controlled conditions for urine collection, some investigators advocate spot or overnight urine collections with normalization of output catecholamines or metanephrines against urinary creatinine excretion [8, 80].
Blood samples should be stored on ice immediately after collection and separated plasma at −80 °C upon analysis. Urine samples should be refrigerated during the collection period while using HCl as a preservative. Urine sample aliquots should be stored frozen at −80 °C to minimize auto-oxidation and deconjugation [53].
When evaluating a patient for possible PHEO/PGL, it is necessary to ask the patient about their current medications. Certain compounds can increase catecholamine levels or interfere with the diagnostic analysis and, thus, result in catecholamine/metanephrine false positives [78]. The major source of interference is tricyclic antidepressants, which can lead to significant elevation of plasma or urinary metanephrines due to inhibition of catecholamine reuptake. Acetaminophen, a drug commonly used for pain and fever, as well as mesalamine and sulfasalazine, can interfere with high-performance liquid chromatographic (HPLC) assays used for measurement of catecholamines [67, 81]. An anxiolytic agent, buspirone, can cause falsely elevated levels of urinary metanephrine in some HPLC assays [67]. Other compounds that may distort catecholamine/metanephrine measurements are listed in Table 12.6.
Localization of Pheochromocytoma
PHEO/PGL localization should only be initiated if clinical evidence for the presence of a tumor is reasonably convincing and biochemical results are strongly positive [28, 59]. If biochemical evidence of a tumor is not compelling, imaging is only justified in patients with a higher probability of tumor development, such as those with hereditary predisposition, previous history of the tumor, or evidence of biochemically silent tumor in carriers with one of the PHEO/PGL susceptibility genes [8, 59]. Based on the tumor biochemical profile, imaging can initially be focused on certain areas of the body. In patients with elevated metanephrine/epinephrine levels, imaging should primarily center on the adrenal gland, as adrenergic phenotype is associated mostly with adrenal tumors. If the scan of adrenal glands is normal, imaging of additional areas of the body should be performed, specifically the abdomen, pelvis, chest, and neck (“eyes to thighs”). A detailed history and careful physical examination may also provide critical information about the possible location of a PHEO/PGL. For instance, postmicturition hypertension suggests urinary bladder PGL [8].
According to expert recommendations, optimal results for PHEO/PGL localization and confirmation are achieved by performing anatomical imaging studies (computed tomography (CT) and magnetic resonance imaging (MRI)) in combination with functional (nuclear medicine) imaging studies [59]. Functional imaging is very useful in detecting primary or metastatic tumors, which could be missed with anatomical modalities [6]. Moreover, imaging plays an important role in the decision-making approach. For example, identification of multiple lesions or metastases before an initial surgery may completely change the treatment plan [8]. An algorithm for localization of PHEO/PGL is depicted in Fig. 12.1. Imaging is also an important part of screening patients with known genetic predisposition to PHEO/PGL development and for follow-up for patients with a history of PHEO/PGL. For genetic mutation carriers, CT or MRI is recommended every few years along with biochemical evaluation. This is particularly important for carriers of the mutations in genes encoding succinate dehydrogenase subunits (SDHx: SDHA, SDHB, SDHC, SDHD) and patients with head and neck PGLs, as these patients often present with biochemically silent tumors [28, 82].
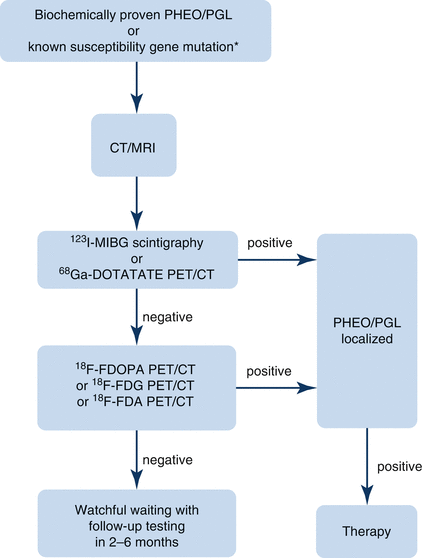
Fig. 12.1
Algorithm for localization of PHEO/PGL. In patients with a biochemically proven PHEO/PGL, as well as in patients with a susceptibility gene mutation known to be associated with a nonsecretory phenotype, anatomic imaging of adrenals/abdomen is suggested. If the result is negative, chest and neck CT or MRI scans should be performed. Afterward, the presence of PHEO/PGL should be confirmed or ruled out with functional imaging [8]. 18 F-FDA 18F-fluorodopamine, 18 F-FDG 18F-fluorodeoxyglucose, 18 F-FDOPA 18F-dihydroxyphenylalanine, 68 Ga-DOTATATE 68Ga-DOTA(0)-Tyr(3)-octreotate, 123 I-MIBG 123I-metaiodobenzylguanidine, CT computed tomography, MRI magnetic resonance imaging, PET positron emission tomography, PGL paraganglioma, PHEO pheochromocytoma
Anatomical Imaging
Anatomical imaging of PHEO/PGL should initially focus on the abdomen and pelvis, followed by the chest and neck if abdominal and pelvic scans are negative [59]. Computed tomography and MRI are widely used in the diagnostic workup for PHEO/PGL, and these modalities have been reported to have similar diagnostic sensitivities [10], although MRI may be superior to CT in detecting extra-adrenal tumors in certain locations (e.g., cardiac) [8]. Ultrasound is not recommended for initial PHEO/PGL localization. Exceptions include ruling out tumors in children and pregnant women, when MRI is not available.
Excellent spatial resolution, wider availability, and a relative low cost suggest a CT scan of the abdomen, with or without contrast, as an initial PHEO/PGL localization method [28]. Computed tomography can be used to localize tumors 1 cm or larger. The sensitivity of CT is approximately 95%, with specificity roughly 70% [83]. Use of intravenous contrast media is preferred to enhance the specificity of the method. However, the CT scan may fail to localize recurrent PHEOs/PGLs because of postoperative anatomical changes and the presence of surgical clips.
An MRI with or without gadolinium enhancement is a very dependable imaging method with sensitivity >95% and specificity similar to CT (70–80%) [10, 84, 85]. MRI has a high sensitivity in detecting adrenal lesions (93–100%) and is a good imaging modality for the detection of intracardiac, juxtacardiac, and juxtavascular PGLs. Moreover, MRI offers feasibility of multiplanar imaging and superior assessment of the relationships between tumor and surrounding vessels. This is important in the evaluation of patients with tumors in the adrenal and cardiac areas and for ruling out vessel invasion. The sensitivity of MRI for detection of extra-adrenal, metastatic, or recurrent PHEOs/PGLs is around 90%. Thus, MRI is preferred in patients with head and neck PGLs and metastatic disease and in patients with CT contrast allergies or in whom radiation exposure is contraindicated (pregnant women, children, patients with known germline mutations, patients with recent excessive radiation exposure) [10, 28].
Functional Imaging
Functional imaging plays an important role in PHEO/PGL workup. Specificity of anatomical imaging studies for PHEO/PGL is not sufficient. Thus, functional studies are needed to confirm the presence of a PHEO/PGL. Functional imaging may also help detect primary and/or metastatic tumors that could be missed on anatomical studies. It is also used to characterize the metabolic activity of the tumors in vivo and for restaging aggressive tumors following treatment completion [86]. Functional imaging studies are enabled by the presence of the cell membrane and/or vesicular catecholamine transport systems in PHEO/PGL cells. Functional imaging modalities used to confirm PHEO/PGL and/or metastatic disease include 123I-metaiodobenzylguanidine (123I-MIBG) scintigraphy, 6-18F-fluorodopamine (18F-FDA), 18F-dihydroxyphenylalanine (18F-DOPA), 11C-hydroxyephedrine, and 11C-epinephrine (not used anymore) positron emission tomography (PET) [87–93]. Currently, these methods are not widely available, and if needed, patients are referred to specialized centers.
In metastatic PHEO/PGL, tumor dedifferentiation may lead to loss of specific neurotransmitter transporters, resulting in difficulties with its localization. In such cases, 18F-fluorodeoxyglucose (18F-FDG) PET imaging or somatostatin receptor scintigraphy may be required. Metastatic PHEOs/PGLs often express somatostatin receptors, which enables somatostatin scintigraphy with the somatostatin analogue octreotide (octreoscan) or DOTA peptides analogues (68Ga-DOTA(0)-Tyr(3)-octreotate (DOTATATE) PET/CT) [94–97].
Metaiodobenzylguanidine Scintigraphy
Historically, functional imaging has been performed using MIBG labeled with radioactive iodine (123I and 131I). MIBG is an aralkylguanidine similar to norepinephrine. 131I-MIBG scintigraphy is not used for imaging because of its longer half-life and lower sensitivity (50%) compared to 123I-MIBG (92–98% in nonmetastatic tumors, 57–79% for metastases) [90, 98–103]. Although the sensitivity of 123I-MIBG for detection of metastases is low, it is very useful in identification of patients who can possibly benefit from palliative treatment with therapeutic doses of 131I-MIBG. Besides PHEOs/PGLs, 123I-MIBG uptake also occurs in other neuroendocrine tumors, such as glomus tumors, carcinoids, or in the sporadic and familial medullary carcinomas of the thyroid. 123/131I-MIBG is physiologically accumulated in the myocardium, spleen, liver, urinary bladder, lungs, salivary glands, large intestine, and cerebellum. Furthermore, in 75% of patients, uptake is shown in normal adrenal glands.
Before MIBG scintigraphy, it is important to withhold the drugs interfering with accumulation of MIBG—it is suggested to do so for 2 weeks prior to exam. Interfering drugs include compounds that deplete catecholamine stores, compounds that inhibit cell catecholamine transporters, and other drugs such as calcium channel blockers or certain α- and β-blockers (Table 12.7) [104–106]. Appropriate blockade with potassium iodide, potassium iodate, 1% Lugol’s solution, or potassium perchlorate is required to prevent an uptake and accumulation of free iodide (123/131I) in the thyroid gland [104].
Table 12.7
Compounds interfering with MIBG uptake by tumors
Interfering agent group | Example | Mechanism of interference | Length of discontinuation before MIBG imaging/treatment |
|---|---|---|---|
Combined α-/β-blocker | Labetalol | MIBG uptake inhibition | 72 h |
Adrenergic neurons blockers | Reserpine, bretylium | MIBG storage depletion | 48 h |
Calcium channel blockers | Amlodipine, diltiazem, nifedipine | MIBG uptake inhibition | 48–72 h |
Inotropic sympathomimetics | Dobutamine, dopamine | MIBG storage depletion | 24 h |
Vasoconstrictor sympathomimetics | Ephedrine, phenylephrine, norepinephrine | MIBG storage depletion | 24–48 h |
β2 adrenoceptor stimulants (sympathomimetics) | Salbutamol, terbutaline, fenoterol | MIBG storage depletion | 24 h |
Other adrenoceptor stimulants | Orciprenaline | MIBG storage depletion | 24 h |
Sympathomimetics for glaucoma | Brimonidine, dipivefrine | MIBG storage depletion | 48 h |
Tricyclic antidepressants | Amitriptyline, clomipramine, nortriptyline | MIBG uptake inhibition | 24–48 h |
Tricyclic-related antidepressants/atypical antidepressants | Maprotiline, trazodone, venlafaxine, mirtazapine | MIBG uptake inhibition | 48 h–8 days |
Antip sychotics (neuroleptics) | Chlorpromazine, haloperidol, perphenazine, risperidone | MIBG uptake inhibition | 24 h–7 days, 1 month for depot forms |
CNS stimulants | Amphetamines, cocaine, caffeine, phenylpropanolamine | MIBG uptake inhibition/MIBG storage depletion/unknown | 24 h–5 days |
Sedating antihistamines | Promethazine | MIBG uptake inhibition | 24 h |
Systemic and local nasal decongestants, compound cough and cold preparations | Pseudoephedrine, phenylephrine, phenylpropanolamine | MIBG storage depletion | 48 h |
Opioid analgesics | Tramadol | MIBG uptake inhibition | 24 h |
Imaging scans are performed at 24 h and again at either 48 or 72 h after injection of the radioisotope, to decipher whether images from earlier scan are tumors or are physiological and fading out.
Positron Emission Tomography
PET has become a more widely available and valuable imaging method, offering high sensitivity, shorter acquisition times, low radiation exposure, and superior spatial resolut ion [86, 107]. Moreover, PET provides a quantifiable estimate of tumor metabolism using standard uptake values (SUV) [86].
Most of the tumors, including PHEO/PGL, exhibit increased glucose metabolism, which enables the use of glucose labeled with 18F (fluoride) for imaging [91, 92]. 18F-FDG PET is highly sensitive for the detection of metastatic PHEO/PGL (approx. 90%), especially in patients harboring SDH subunit B (SDHB) gene mutations. Sensitivity of 18F-FDG PET for detection of primary, nonmetastatic PHEOs/PGLs is 88%, with a specificity similar to that of 123I-MIBG. Thus, 18F-FDG PET/CT is recommended for localization of metastatic disease [28, 108].
The majority of the radiopharmaceuticals used for PET detection of PHEO/PGL enter the tumor cell using the cell membrane norepinephrine transporter. A positron emitting analogue of dopamine, 6-18F-FDA, is a very useful sympathoneuronal PET imaging agent for catecholamine-synthesizing cells [109]. 18F-FDA PET has a high sensitivity for both primary PHEOs/PGLs and metastases (77–100% and 77–90%, respectively), with specificity more than 90% [100, 108]. Unfortunately, 18F-FDA PET/CT is not yet widely available. Other PET imaging tracers, 11C-hydroxyephedrine and 11C-epinephrine, have been shown to only have limited application in diagnostic imaging because of the short half-life of 11C (20 min) [110, 111].
18F-DOPA is an amino acid analogue and catecholamine precursor that is taken up by the amino acid transporter [112]. Pretreatment with carbidopa enhances tumor uptake of tracer and improves sensitivity due to inhibition of DOPA decarboxylase [113]. 18F-DOPA is extremely sensitive (81–100%) for the localization of nonmetastatic PHEO/PGL and head and neck PHEO/PGL [87, 108, 114, 115]. However, for detection of metastatic and SDHB mutation-related PHEOs/PGLs, sensitivity is not satisfactory (45% and 20%, respectively) [8].
Somatostatin Receptor-Based Imaging
Somatostatin receptors are expressed in up to 73% of PHEO/PGL cells in vitro [116], and scintigraphy using octreotide (111In-pentreotide) has been used for PHEO/PGL localization, specifically for localization of head and neck PGLs. However, the sensitivity of this imaging modality is low, and thus, it is inferior to 123I-MIBG scintigraphy [117]. Still, octreoscan can be useful in detection of tumors that express somatostatin receptors and are negative on other scans [118].
For somatostatin receptor-based PET/CT imaging , radiolabeled DOTA peptide analogues (DOTATATE, DOTATOC, and DOTANOC) were shown to be superior to all other imaging methods. For instance, 68Ga-labeled DOTA peptides have been found to be highly sensitive for localization of neuroendocrine tumors, including PHEO/PGL [96, 97, 119–126]. In recent studies from Janssen et al. [94, 95], 68Ga-DOTATATE PET/CT was shown to be clearly superior to all other functional imaging modalities, including 18F-FDG PET/CT, 18F-FDOPA PET/CT, and 18F-FDA PET, for localization of both sporadic and SDHB-related metastatic PHEO/PGL. The costs of imaging based on radiolabeled DOTA peptides are comparable to 18F-FDG PET or 123I-MIBG scintigraphy. These new modalities are expected to be more broadly available in few years.
Genetic Testing
From a clinical point of view, it is necessary to consider genetic testing in addition to diagnostics and appropriate therapy, especially in patients with a suspected hereditary form of the disease and in their first-step relatives. If the PHEO/PGL susceptibility germline mutation is present, patients need to be screened regularly, even if the disease is not obvious. Particular gene mutations present with a specific clinical and biochemical phenotype. Early identification of a mutation allows a physician to predict the course of disease, risk of malignancy, and heritability and helps to choose an appropriate treatment strategy. In order to diagnose PHEO/PGL and identify a specific mutation, it is necessary to take a comprehensive personal and family history, perform meticulous clinical and biochemical examinations, and use adequate imaging methods. In PHEO/PGL, it is important to particularly assess the location of a tumor, biochemical phenotype, age of a patient, and the presence of any tumors besides PHEO/PGL [6].
Hereditary disease should be su spected in patients from families with two or more cases of PHEO/PGL or with syndromes associated with PHEO/PGL. In syndromic forms of PHEO/PGL, underlying mutations can be predicted based on a combination of characteristic tumor types in the patient or their family members (e.g., renal cell carcinoma and hemangioblastoma or retinal angioma in von Hippel-Lindau (VHL) disease, medullary thyroid cancer in multiple endocrine neoplasia type 2) or based on characteristic clinical phenotype (e.g., café au lait spots and eye symptoms—Lisch nodules, optical gliomas—in neurofibromatosis type 1, polycythemia in mutations in hypoxia-inducible factor 2α (HIF2A) gene, Hirschsprung’s disease in multiple endocrine neoplasia type 2) [127, 128]. Hereditary PHEO/PGL syndromes, associated tumors, and other characteristics associated with various mutations are in Table 12.5.
From a biochemical point of view, adrenergic mixed phenotype is observed in PHEOs/PGLs associated with NF1 (neurofibromin 1), RET (rearranged during transfection), KIF1Bβ, and MAX (myc-associated factor X) mutations. TMEM127 (transmembrane protein 127)-mutated tumors present with high metanephrine concentrations, which means they are solely adrenergic [50, 129]. PHEOs/PGLs associated with mutations in VHL, SDHx, SDHAF2 (SDH assembly factor 2), HIF2A, FH (fumarate hydratase), IDH (isocitrate dehydrogenase), and PHD1/2 (HIF prolyl hydroxylase domain-containing protein 1/2) genes are mostly extra-adrenal, except for VHL-mutated tumors [50, 130–132], and usually present with dopaminergic and/or noradrenergic biochemical phenotype [54, 133, 134]. VHL– and HIF2A-mutated PHEOs/PGLs typically exhibit a noradrenergic phenotype, and SDHx tumors are associated with a dopaminergic component. Rarely, they are biochemically silent [50, 129, 133, 134] (Fig. 12.2).
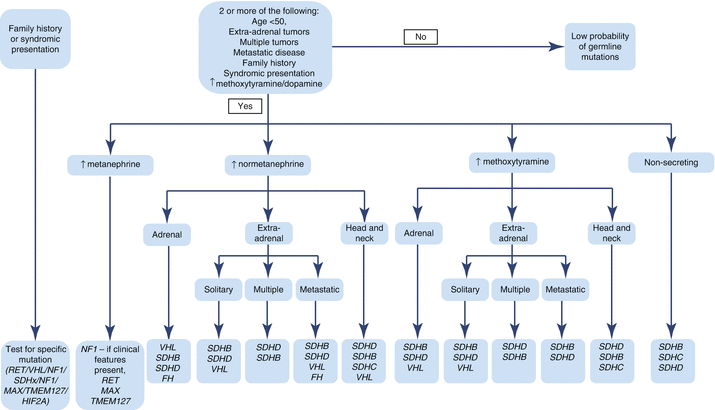
Fig. 12.2
Algorithm for genetic testing based on biochemical phenotype and clinical presentation of PHEO/PGL. In patients with a known family history of PHEO/PGL susceptibility mutation, this mutation should be tested first. Patients presenting with features of one of the syndromes associated with PHEO/PGL (NF1, VHL, MEN2, Pacak-Zhuang, etc.) should be tested for genetic mutations linked to those syndromes. In NF1, diagnosis is usually made on clinical presentation alone, and no genetic testing is needed. In patients without a known family history of PHEO/PGL, the direction of genetic testing is based upon the biochemical profile and localization of the tumor. For some of the known genetic mutations, biochemical profile and/or tumor location has not yet been elucidated (Table 12.5) [6, 135–137]. FH fum arate hydratase, HIF2A hypoxia-inducible factor 2α, MAX myc-associated factor X, MEN2 multiple endocrine neoplasia, type 2, NF1 neurofibromin 1, NF1 neurofibromatosis type 1, PGL paraganglioma, PHEO pheochromocytoma, RET rearranged during transfection proto-oncogene, SDHA, SDHB, SDHC, SDHD succinate dehydrogenase subunits A, B, C, and D, SDHx succinate dehydrogenase complex gene mutations, TMEM127 transmembrane protein 127, VHL von Hippel-Lindau
Presence of metastases in PHEO/PGL patient leads to suspicion of SDHB or FH mutations. Familial forms of PHEO/PGL usually show autosomal dominant inheritance, which means that children of mutation carriers have a 50% chance of inheriting the mutation from a parent. However, we cannot forget about the low penetrance of PHEO/PGL in some mutations (e.g., SDHx)—in these cases, a negative family history does not rule out the presence of a familial form of PHEO/PGL. In SDHD, SDHAF2, and MAX mutations, the risk of disease depends on which parent is the mutation carrier. The disease will only develop if the mutated gene is inherited from the father [138]. Germline mutations are present in 8–24% of apparently sporadic PHEOs/PGLs. These mutations often appear as de novo mutations or are associated with low penetrance [127, 128, 139]. In these patients, an underlying gene mutation should be considered if the tumor is extra-adrenal and malignant or if the diagn osis of PHEO/PGL is made at an early age [136].
Management of Patient with PHEO/PGL
Appropriate management of patients with PHEO/PGL requires a close collaboration of several specialists, including an endocrinologist, internist, radiologist, anesthesiologist, surgeon, and, if needed, oncologist [10, 59]. Currently, the only available curative treatment for PHEO/PGL is surgery. Thus, the optimal therapy for PHEO/PGL is a prompt, ideally complete, surgical removal of the tumor to prevent potentially life-threatening complications. Surgical debulking or extensive metastases removal may also allow for long-term remission in patients with locoregional or isolated resectable distant metastases, or it can palliate symptoms related to tumor mass or catecholamine excess [140]. Systemic chemotherapy and radiotherapy are possible treatment options for patients who are not surgical candidates, although these have only palliative character. However, recent progress in understanding the molecular mechanisms involved in PHEO/PGL development has driven introduction of new promising therapeutic options.
Medical Management and Preparation for Surgery
Immediately after diagnosis, all patients with biochemically active PHEO/PGL should be placed on sufficient adrenoceptor blockade to control symptoms and reduce the risk of hypertensive crises and organ damage mediated by the effects of released catecholamines [6]. There is no consensus regarding the drugs recommended for preoperative management because of wid e-ranging practices, international differences in available or approved therapies, and a lack of studies comparing different medications. However, α-adrenoceptor antagonists, calcium-channel blockers, and angiotensin-receptor blockers are recommended [28, 141]. Drugs that can be used in symptom management and presurgical blockade, with suggested doses, are listed in Table 12.8.
Table 12.8
Drugs used for symptom management and presurgical blockade in patients with PHEO/PGL
Drug | Classification | Suggested dose | Use | Common side effects |
|---|---|---|---|---|
α-Adrenoceptor blockers | ||||
Phenoxybenzamine | Long lasting, irreversible, noncompetitive | 10 mg 1–3 times daily | First choice for α-adrenoceptor blockade | Orthostatic hypotension, nasal congestion, tachycardia, dizziness |
Prazosin | Short acting, specific, competitive | 2–5 mg 2–3 times daily | – When phenoxybenzamine is not available – For patients who do not tolerate phenoxybenzamine – For patients with mild hypertension | |
Terazosin | Short acting, specific, competitive | 2–5 mg daily | ||
Doxazosin | Short acting, specific, competitive | 4–24 mg 2 times daily | ||
β-Adrenoceptor blockers | ||||
Atenolol | Cardioselective | 12.5–25 mg 2–3 times daily | To control tachyarrhythmia resulting from catecholamine excess or from α-adrenoceptor blockade | Fatigue, dizziness, exacerbation of asthma |
Metoprolol | Cardioselective | 25–50 mg 3–4 times daily | ||
Propranolol | Nonselective | 20–80 mg 1–3 times daily | ||
Calcium channel blockers | ||||
Amlodipine | 10–20 mg daily
Related posts: Pheochromocytomas and Paragangliomas: Genetics and Pathophysiology Pheochromocytomas and Paragangliomas: Genetics and Pathophysiology
 Adrenal Zonation and Development Adrenal Zonation and Development
 Diagnosis and Management of Adrenal Insufficiency
Pheochromocytomas and Paragangliomas: Genetics and Pathophysiology Diagnosis and Management of Adrenal Insufficiency
Pheochromocytomas and Paragangliomas: Genetics and Pathophysiology
 Genetics and Pathophysiology of Congenital Adrenal Hyperplasia Genetics and Pathophysiology of Congenital Adrenal Hyperplasia
 Adrenal Cushing’s Syndrome: Updates on Overt and Mild Hypercortisolism Adrenal Cushing’s Syndrome: Updates on Overt and Mild Hypercortisolism
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

| |||