Syndrome
Gene (chromosome)a
Clinical manifestations
% with PHEO
% PHEO bilateral
References
von Hippel Lindaub
VHL (3p25-p26)
PHEO, cerebellar hemangioblastoma, retinal angiomas, renal cell carcinoma, pancreatic cysts, pancreatic NETs in 5–10%
10–20%
40–80
[11]
Multiple endocrine neoplasia type 2Ac
Multiple endocrine neoplasia type 2B
RET (10q11.2)
PHEO, MTC, hyperparathyroidism
PHEO, MTC, mucosal neuromas, colonic ganglioneuromatosis, Marfanoid facies, skeletal abnormalities
50%
50–80
[10]
von Recklinghausen’s disease
NF1 (17q11.2)
Neurofibromata, >6 café au lait spots, axillary freckling, skeletal abnormalities and PHEO
5–10%
10
[15]
PGL-1
SDHD (11q23)
Parasympathetic head and neck PGL ± PHEO
Uncommon
–
[14]
PGL-2
SDHAF2 (11q13.1)
Parasympathetic head and neck PGL (non-functioning)
Not described
–
[38]
PGL-3
SDHC (1q21)
Parasympathetic head and neck PGL, PHEO rare
Rare
–
[12]
PGL-4
SDHB (1p35-p36)
Sympathetic abdominal PGL and PHEO—50% malignant
PGL or PHEO in 70%
[13]
Familial pheochromocytoma
TMEM127 (2q11)
PHEO
?
43
[40]
Familial neural crest-derived tumours
KIF1B (1p36.2)
PHEO or neuroblastoma
?
?
[102]
Aetiology and Molecular Genetics
The molecular basis of sporadic catecholamine-secreting tumours is poorly understood and few somatic mutations have been identified in tumour specimens. However, our understanding of the molecular basis of tumorigenesis in familial pheochromocytoma continues to develop. Experimental evidence suggests that alterations in the genes responsible for these familial pheochromocytoma (VHL, RET, NF1 and SDHx) impair c–Jun dependent neuronal apoptosis during normal development. This unifying explanation is known as the ‘developmental culling hypothesis’ [20, 21]. The individual genetic disorders giving rise to PHEO and PGL are discussed below.
Von Hippel–Lindau Disease (VHL)
This autosomal-dominant disease is a significant cause of PHEO presenting in children with an incidence of 1 per 36,000 live births [22]. VHL is characterised by an increased risk of hemangioblastoma of the CNS and retina, renal cell cancer (RCC), neuroendocrine tumours of the pancreas, endolymphatic sac tumours and PHEO. 10–20% of affected patients develop PHEO and the tumours are often bilateral. Multiple cysts of the pancreas, kidney and epididymis are also common [23].
There are four subtypes of VHL: Types 1, 2A, 2B and 2C. All subtypes are due to germ-line abnormalities of the VHL gene on chromosome 3p25 [24]. There is a strong genotype–phenotype correlation. Type 1 disease is due to truncating VHL mutations that are inactivating and lead to loss of function. Type 1 disease is not associated with PHEO. Each of the Type 2 diseases is due to specific missense activating mutations and leads to gain of function of the VHL protein. Each variant has a specific tumour-risk pattern. Type 2A has an increased risk of retinal and CNS hemangioblastoma and PHEO but not RCC, Type 2B has an increased risk of RCC, hemangioblastoma and PHEO; and Type 2C has an increased risk of PHEO only [24, 25].
Nearly, all VHL families harbour specific germ-line mutations, but up to 20% of cases of VHL disease occur de novo [26]. Although malignant PHEO is reported in less than 5% of patients with VHL, those patients with subtypes that lead to RCC and hemangioblastoma have a reduced life expectancy and should be screened from 5 years of age onward with MRI of the brain and spinal cord, kidneys and adrenal glands [27]. Patients with VHL who are undergoing a surgical procedure should have preoperative evaluation of urine or plasma metanephrines to exclude occult PHEO.
Multiple Endocrine Neoplasia Type 2A and 2B (MEN 2A and 2B)
MEN 2 is an autosomal dominantly inherited endocrine tumour syndrome that affects 2–3 per 100,000 of the population. It is due to activating germ-line mutations of the gene on chromosome 10q11 that encodes the RET tyrosine kinase receptor [10]. Two main clinical subtypes are recognised, MEN 2A and MEN 2B.
MEN 2A is characterised by medullary thyroid cancer (MTC) in all patients, hyperparathyroidism in 30% and PHEO in 50%. MEN 2B is much less common accounting for only 10–15% of patients with MEN. The clinical features of MEN 2B include Marfanoid facies, skeletal deformities, mucosal neuromas of the tongue and lips, ganglioneuromatosis of the intestine, MTC and PHEO. A third disorder, familial MTC (FMTC), is also due to RET mutations but not associated with PHEO. Although MTC is almost always the initial presenting disorder in MEN2, as many as one-third of patients may present with MTC and PHEO simultaneously and so the latter should always be excluded prior to surgery for MTC [28].
The clinical features of the disease (phenotype), age of onset and aggressiveness of MTC are dependent upon the specific codon mutation inherited by the patient (genotype) [27, 29]. Patients with MEN 2A have mutations that affect the extracellular ligand-binding domain of the receptor (exons 10 and 11 of the RET gene) and 85% of patients have codon 634 RET mutations. The remaining 10–15% have mutations of codons 609, 611, 618 and 620 [30]. In contrast, those with MEN 2B have mutations affecting the intra-cellular, tyrosine kinase domain (exons 15 and 16) and 95% of patients have the codon 918 mutations, or more rarely codon 883 mutation [30]. Patients with FMTC have mutations affecting codons 768, 790, 791, 804 and 844 (exons 13 and 14) although mutations are described in exons 10 and 11, denoting an overlap between FMTC and MEN 2A [10]. Interestingly, although PHEO is observed in 50% of individuals with codon 634 and 918 mutations, it occurs rarely in those with mutations in exon 10 (codons 609, 611, 618 and 620) or exon 15 (codon 791 and 804) [31, 32].
Neurofibromatosis Type 1 (Von Recklinghausen Disease)
Neurofibromatosis type 1 (NF-1) is an autosomal-dominant condition that occurs in 1 in 3000 live births and is due to mutations in the NF1 gene on chromosome 17q. Half represent de novo changes [24]. PHEO develops in 5% of those affected, usually in adulthood [33]. Diagnosis of NF-1 is usually straightforward because of the presence of cutaneous neurofibromata, café au lait spots and axillary and inguinal freckling. Genetic testing is problematic because the gene contains 60 exons with numerous pseudogenes [34]; however, it is rarely necessary due to the obvious clinical signs. Screening for PHEO should be performed in individuals with the disorder who develop hypertension.
Hereditary Paraganglioma and Pheochromocytoma Syndromes
Four inherited syndromes, PGL-1, PGL-2, PGL-3 and PGL-4 syndromes, have varying predisposition to development of PHEO, extra-adrenal sympathetic PGL and parasympathetic head and neck PGL. These syndromes are caused by germ-line mutations of genes encoding subunits of the mitochondrial complex II enzyme, succinate dehydrogenase (SDH), which is involved in the tricarboxylic acid cycle. PGL-1 syndrome is caused by SDHD mutations, PGL-3 syndrome is caused by SDHC mutations and PGL-4 syndrome is caused by SDHB mutations. SDHD mutations are also found in up to 11% of sporadic PHEO [35] and in up to 50% of sporadic head and neck PGL tumours [36]. These SDHD mutations have an unusual paternal origin of inheritance [37]. PGL-2 syndrome is due to mutations of the SDHAF2 gene which encodes a protein required for flavination of the SDHA subunit [38].
PGL-1 syndrome is associated with parasympathetic head and neck PGL (chemodectoma or glomus tumour) in middle-aged patients and occasionally PHEO [14, 36]. PGL-2 is the least common syndrome and is associated with parasympathetic head and neck PGL. PGL-3 syndrome is rare with only 10 families in the literature. The disorder is associated with development of parasympathetic head and neck PGL only [36]. PGL-4 causes predominantly abdominal sympathetic PGLs and occasionally PHEO [13], but almost half are malignant [39]. Tumours may develop from the first decade of life. Diagnosis of initial tumour is at a median age of 32 years and there is an estimated penetrance of 45% by age 40 [36].
Other Genetic Disorders
Germ-line inactivating mutations of the transmembrane-encoding gene TMEM127 on chromosome 2q11 have been shown to be associated with the development of PHEO [40]. Like RET, TMEM127 acts as a tumour suppressor gene. Large-scale mutational analysis of 990 patients with sporadic PHEO revealed TMEM127 mutations in just under 2%, with bilateral tumours in over a third of these individuals [41].
The Carney triad of gastro-intestinal stromal tumours (GISTs), pulmonary chondroma and PHEO/PGL occurs predominantly in women and is most commonly associated with deletions of chromosome 1pcen13-q21 which contains the SDHC gene although the exact genetic defect is unknown [42]. However, a separate condition, the Carney-Stratakis syndrome of GIST and PHEO/PGL, has been demonstrated to be due to SDHB, SDHC and SDHD mutations in a proportion of patients [36].
Genetic testing should be performed in all children and adolescents that present with PHEO or PGL. The precise order of genetic testing is dependent upon the clinical presentation. Thus, patients presenting with PHEO and MTC should undergo screening of RET, whereas those with isolated PHEO should undergo VHL and if negative, SDHB, SDHD and SDHC testing. Individuals with extra-adrenal tumours in the abdomen and chest should undergo VHL, SDHB, SDHD and SDHC screening, whereas those with head and neck PGL are most likely to harbour mutations in SDHD, SDHB, SDHC, SDHAF2 and occasionally the VHL gene. Counselling and testing of first-degree relatives should follow the detection of index cases.
Pathology
Grossly pheochromocytomas are soft vascular tumours that when sliced are a pink/tan colour. Tumours are usually less than 5 cm in maximal diameter but can be much larger. Microscopically, tumours have a regular nested (Zellballen) pattern of polyhedral cells which stain yellow with chromic acid [43] (hence the term ‘chromaffin positive’) and contain large nuclei with numerous secretory granules. A layer of sustentacular cells surrounds the nests of chromaffin-positive cells. These cells are more prominent in benign tumours [44, 45] and stain positive for the immunohistochemical marker S-100. Tumours also demonstrate immunoreactivity for chromogranin A and synaptophysin [46–48].
The incidence of malignant PHEO is around 12% in children [5], but is higher in extra-adrenal sympathetic tumours, particularly those due to SDHB mutations. Only distant metastases and local invasion are definitive markers of malignant tumours. However, a number of surrogates suggest an increased risk of malignancy: tumour diameter >5 cm (75% prevalence of malignancy) [5]; presence of lymphovascular or capsular invasion; presence of confluent tumour necrosis; increased Ki-67 proliferation index and absent S-100 staining of sustentacular cells [45, 49–51]. Scoring systems to predict malignancy have been developed (PHEO of the Adrenal Gland Scaled Score or PASS) [52] using some of the above criteria and other histological appearances including the presence of high cellularity, tumour cell spindling, cellular monotony, increased mitotic figures, atypical mitotic figures, nuclear pleomorphism and cellular hyperchromasia. However, significant inter- and intra-observer variability of PASS component scoring highlights the limitations of current scoring systems in diagnosing malignancy [53].
Malignant tumours have increased tumour expression of some biological markers [telomerase catalytic subunit hTERT, heat shock protein (HSP) 90, cyclo-oxygenase, N-cadherin, vascular endothelial growth factor (VEGF)] and decreased expression of others (neuropeptide Y (NPY) and EM66) [54]. The use of these indicators to predict malignant behaviour is not routine in clinical practice.
Finally, SDHB protein expression by tumour immunohistochemistry is absent in all PHEO/PGL presenting in patients affected by SDHB, C and D mutations, but present in all those presenting as part of MEN2, VHL and NF1 and in almost 90% of those with no germ-line mutation [55]. SDHB immunohistochemistry could be a useful to guide genetic testing and, in the case of SDHB-related PHEO/PGL, assist in identifying tumours with high malignant potential.
Clinical Presentation
Sustained hypertension is the most common presenting finding in children (Table 8.2) in contrast to adults who often have the classic triad of episodic sweating, palpitations and headache [19, 56]. In addition to signs and symptoms of excess catecholamines, tumours may present with local mass effects (abdominal pain with or without abdominal distension), or as an incidental finding on imaging studies, or during biochemical screening tests. A feature sometimes noted in tumours related to VHL and MEN 2 syndromes is the lack of symptoms. This may be that this is due to ascertainment bias (e.g. tumours detected on screening do not reach a size where they become symptomatic) rather than a feature of genetic tumours per se [57].
Table 8.2
Clinical presentation of pheochromocytoma and functioning paraganglioma in children compared with adults
Children with PHEO/PGL | Adults with PHEO/PGLa | |
|---|---|---|
Age | 11 years | |
Male: Female | 1.6:1 | 1:1 |
Presenting symptom | ||
Sustained hypertension | 90–100 | 68 |
Headache | 81–95 | 90 |
Palpitations | 35–63 | 72 |
Sweating | 69–90 | 92 |
Mass effect | 30–38 | ? |
Evaluation
The approach to diagnosis in children is summarised in Fig. 8.1 with additional explanations in the following sections.
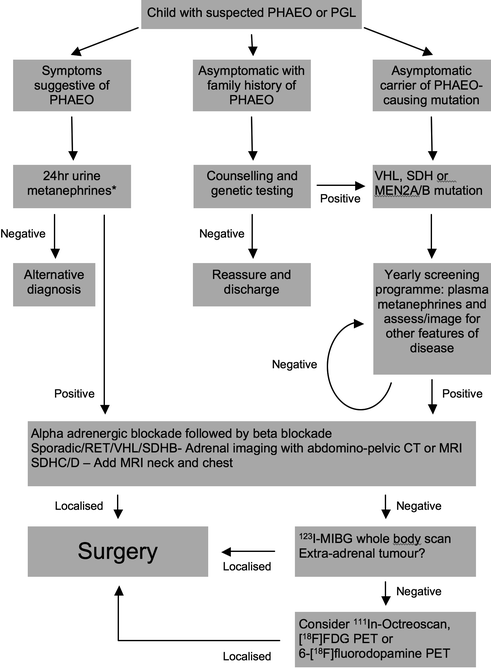
Fig. 8.1
Suggested algorithm for the investigation of a child with suspected pheochromocytoma or paraganglioma (PHEO pheochromocytoma, PGL paraganglioma, see text for other abbreviations)
History
On presentation, the child and parents should be questioned about the presence and duration of symptoms relating to catecholamine excess such as headache, palpitations, panic attacks, sweating and about neurological symptoms such as blurred vision and weakness. In addition, specific questioning should investigate the possibility of other associated conditions such as thyroid nodules, hypercalcaemia, megacolon and renal tumours. Family history of PHEO, PGL and any related endocrine tumour (see Table 8.1) should be noted along with a pedigree of affected and unaffected family members. Finally, a full past medical history of medical problems and surgical procedures that might impact anaesthesia or operation should be documented.
Physical Examination
The most common abnormality on physical examination is hypertension but the physical examination is often normal. However, physical exam is diagnostic in patients with NF-1 (multiple cutaneous neurofibromata, café au lait spots and axillary/inguinal freckling) and in patients with MEN 2B (Marfanoid appearance, lingual neuromas, signs of Hirschsprung’s megacolon, neck scar from previous thyroid surgery). Adrenal and extra-adrenal tumours are not usually palpable unless they are very large, although tumours in the neck and at the abdominal aortic bifurcation may be palpable.
Investigations
The presence of catecholamine-secreting tumours is confirmed by the biochemical testing of plasma or urine for fractionated catecholamines (adrenaline, noradrenaline and dopamine) or metanephrines (metadrenaline, normetadrenaline and 3-methoxytyramine). When catecholamine excess is determined then imaging is done to localise the tumour.
The majority of children with PHEO, particularly if symptomatic, will have increased levels of urinary catecholamines and metanephrines. Metanephrines result the enzyme catecholamine-O-methyltransferase (COMT) within the tumour metabolises catecholamines. Plasma and urinary metanephrines are more sensitive (99 and 97% respectively) than plasma and urinary catecholamine (86 and 84% respectively) because catecholamine secretion may be intermittent [58, 59]. Therefore, the most appropriate tests in children are urinary metanephrines or if urine collection is not feasible then plasma metanephrines. The increased sensitivity of plasma metanephrines is also valuable to detect early disease when there is a known genetic mutation that puts a patient at risk for PHEO. Elevation of metanephrines greater than 4× the upper limit of the reference range is 100% diagnostic for a chromaffin tumour [60]. Metanephrine estimations may be affected by certain drugs (e.g. monoamine oxidase inhibitors, paracetamol, tricyclic antidepressants, sympathomimetics such as ephedrine and phenylephrine, amphetamines and levodopa) so these drugs should be discontinued prior to testing [61].
Tumours may have characteristic biochemical secretions and are classified as either noradrenergic or adrenergic. For example, PGLs and PHEOs arising in association with VHL disease are noradrenergic and predominantly secrete noradrenaline and normetadrenaline [62]. This is likely due to VHL tumours having reduced expression of the enzyme phenylethanolamine-N-methyltransferase (PNMT), which converts noradrenaline to adrenaline [63]. Adrenergic PHEOs secrete a mixture of adrenaline/metadrenaline and noradrenaline/normetadrenaline. They may be sporadic or due to RET and NF1 mutations. Dopamine and its metabolite 3-methoxytyramine are occasionally the predominant secretions, usually from PGL arising due to SDHx mutations. Such tumours are often asymptomatic.
Differential Diagnosis
Definitive biochemical findings of elevated catecholamines denote a PHEO or a PGL. In young children, neuroblastoma is a possibility, but may be distinguished from PHEO and PGLs because neuroblastomas do not secrete physiologically active catecholamines [19]. PHEO or PGL require preoperative alpha- and beta- adrenergic receptor blockade so it is important to distinguish these tumours from neuroblastomas. Therefore, in addition to VMA and HVA, children that present with a diagnosis of neuroblastoma and symptoms of catecholamine excess should have 24-h urinary measurements of fractionated catecholamines and metanephrines.
Imaging and Localisation
Computerised Tomography (CT) and Magnetic Resonance Imaging (MRI)
Radiological investigations to localise the tumour should only be performed after a confirmed biochemical diagnosis. Abdominal and pelvic CT and MRI have a high sensitivity (90–100%) and specificity (70–80%) [64, 65] and will detect the majority of tumours, particularly if they are symptomatic. However, CT and MRI may lack the sensitivity to localise tumours <1 cm diameter that may be detected by early biochemical screening. Contrast CT is quicker and may be more readily available, but requires exposure to radiation. Ionic contrast has been reported to precipitate catecholamine release from PHEO and ‘PHEO crisis’, but non-ionic contrast is safe [66]. MRI (Fig. 8.2) avoids radiation and can distinguish PHEO (which appear hyperintense on T2-weighted images) from other types of adrenal mass (usually hypointense) [67] but may require general anaesthetic in children. Ultrasound may be useful for surveillance of the neck in children at risk of head and neck PGL. Extra-adrenal PGL may occur in the head and neck (5%), the bladder (10%), along the sympathetic chain in the thorax (10%), and along the sympathetic chain in abdomen (75%), either above (juxtarenal) or below (organ of Zuckerkandl), the origin of the inferior mesenteric artery [68]. Due to the increased likelihood of extra-adrenal and multi-focal disease in children, particularly those with familial disease secondary to SDHx mutations, extra-adrenal sites often require investigation.
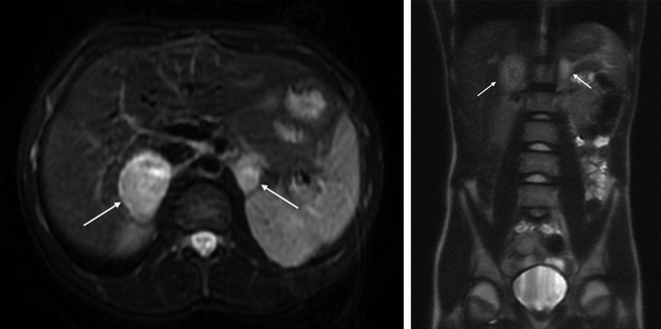
Fig. 8.2
Axial (left) and coronal (right) images from the MRI abdomen of a 13-year-old boy with VHL disease. Bilateral PHEO were diagnosed on biochemical screening. Note the hyperintense appearance of the tumours (white arrows) on axial T2-weighted images. He was treated with laparoscopic adrenalectomy for the 45-mm right-sided tumour and laparoscopic cortical-sparing subtotal adrenalectomy for the 20-mm left-sided tumour, performed sequentially
Other Imaging and Investigations
If initial imaging is negative or reveals extra-adrenal disease, functional imaging with 123I-MIBG (Meta-iodobenzylguanidine) or 111In-octretide may identify occult or multi-focal tumours, confirm functionality of extra-adrenal lesions and detect metastatic disease. Radiolabelled MIBG is taken-up by chromaffin cells via the vesicular monoamine transporter 1 (VMAT-1) because it resembles noradrenaline. It is 80–90% sensitive and more than 95% specific [67]. Radiolabelled octreotide binds to somatostatin receptors commonly expressed by adrenergic tissue and is 50–70% sensitive. Although less sensitive than MIBG, octreotide scanning may be useful if cross-sectional imaging and MIBG scanning are negative. Routine use of functional scans in the presence of adrenal lesions seen on CT/MRI may lead to false-positive localisation of other non-catecholamine-secreting incidentalomas [69, 70], so their use is best reserved for the detection of occult or extra-adrenal disease or for post-operative investigation of residual or metastatic disease. Various forms of positron emission tomography (PET) scanning have shown promise. In one study, imaging with 6-[18F] fluorodopamine was 98% sensitive and 100% specific and was superior to 123I-MIBG [71]. Factors predicting MIBG negativity were young age, hereditary aetiology and lack of VMAT-1 expression in tumours. It is therefore likely that 6-[18F]fluorodopamine PET will be of use in detecting PGL in young patients when conventional imaging and MIBG have failed. It should also be considered for locating metastatic disease [2]. Finally, selective adrenal vein sampling has proved misleading in PHEO and has very limited value [69].
Management
Biochemical diagnosis and localisation of PHEO or PGL should be followed by medical preparation to control blood pressure and then prompt surgical excision.
Preoperative Management
Major operations done in the presence of undiagnosed PHEO are associated with a mortality of between 25 and 100%; therefore, preoperative preparation to protect against catecholamine excess and triggers of secretion such as induction of anaesthesia are essential for a successful outcome. Although practice varies, alpha-adrenergic blockade, with or without beta-adrenergic blockade, is the most common strategy. In the author’s unit, supervised initiation of alpha blockade using oral phenoxybenzamine (0.25–1.0 mg/Kg/day in 3 divided doses) to control blood pressure is the first step. Once alpha blockade is established (usually requiring 4–7 days of therapy) then beta blockade to control tachycardia is commenced (e.g. propranolol 0.25–0.5 mg/Kg/day in 3 divided doses). This sequence of alpha blockade first and then beta blockade is important to avoid dangerous elevations in blood pressure that occur with alpha-induced vasoconstriction. Pharmacological relief of chronic vasoconstriction allows restoration of the inevitable associated depleted intravascular volume over 2–4 weeks. In the week prior to surgery, patients are admitted for supervised blood pressure monitoring and further optimisation of alpha and beta blockade to ensure a goal of a normal resting blood pressure and a postural drop in blood pressure without the normal compensatory tachycardia. At this point it is safe to proceed with an operation. Other regimens have been described, e.g. alpha blockade with doxazosin or prazosin, with and without beta blockade [72], using calcium channel blockers alone [73], and using the catecholamine synthesis blocker, metyrosine, if there are concerns about cardiac failure [74]. However, it must be stressed that preparation should be carried out by a multi-disciplinary team that is experienced in managing such patients and that the familiarity and experience of the team with the condition is probably more important than the exact regimen.
Surgery
Again, a team approach, involving personnel experienced in the surgery for PHEO/PGL, is crucial to ensure a successful outcome in children with the disease. Surgeons experienced in treating such patients will counsel children and their families about possible associated genetic disorders, surgical approach, possible steroid dependence post-operatively, and the need for long-term follow-up. PHEO and intra-abdominal PGL account for 95% of these tumours and can be dealt with by laparotomy or laparoscopic surgery.
Adrenalectomy for PHEO
Due to the rarity of PHEO in children, there are no randomised trials concerning the most appropriate method of surgical excision; however, one small, randomised study in adults reported reduced blood loss, operative time and length of hospital stay with laparoscopic resection [75]. The only disadvantage observed was an increase in intraoperative haemodynamic instability, which has been reported previously [76] but it is usually controlled without significant problems. There are several case series of adults comparing laparoscopic and open adrenalectomy for a variety of adrenal pathologies that favour the laparoscopic approach in terms of blood loss, post-operative pain, length of hospital stay, earlier return to alimentation and earlier return to normal activity [77–85]. Laparoscopic adrenalectomy has also been demonstrated to be safe in children [86] and is an accepted approach for unilateral and bilateral adrenal pathology.
Controversial issues regarding surgery for PHEO include the need of unilateral versus bilateral adrenalectomy for children with a high risk of bilateral disease when imaging has detected only one involved gland and the use of cortical-sparing resections during bilateral adrenalectomy in order to avoid long-term steroid dependence. Follow-up data suggest that unilateral laparoscopic adrenalectomy is the best approach for disease that is unilateral on imaging, followed by regular follow-up with plasma metanephrine measurements for early detection of contralateral disease [87]. Cortical-sparing surgery has been reported in retrospective studies of selected adult patients but it is probably only feasible with small tumours (e.g. <26 mm) [88]. Because a small amount of adrenal medulla is inevitably left behind with the preserved cortex there is a significant risk of recurrent disease (10–38%) [89–92]. The risk of recurrence must be carefully weighed against the substantial morbidity associated with adrenal insufficiency and Addisonian crises that 20% of children will suffer following bilateral adrenalectomy [92].
Following laparoscopic surgery, post-operative stay is <48 h for unilateral surgery but will be longer for bilateral surgery, where pain control and commencement of steroid replacement regimens tend to increase hospital length of stay.
Surgery for Thoracic and Abdominal PGL
Tumours along the sympathetic chain can be technically challenging due to their close relationship to the great vessels and visceral artery branches. For this reason, minimally invasive surgery may not be feasible, and thoracotomy and laparotomy may be the preferred options.
Surgery for Malignant PHEO
The presence of metastatic disease and local invasion is the only definite signs of malignant pheochromocytoma, but size >5 cm and history of genetic disease (SDHB mutations) are associated with increased risk. The choice between laparoscopic and open surgery will therefore depend upon the presence of family history combined with appearances on preoperative imaging. Large tumours with evidence of renal, vena caval, duodenal or hepatic involvement on the right side, or pancreatic, splenic, renal or colonic involvement on the left side, and/or local nodal metastases should be treated with open radical adrenalectomy and even more radical local excisions if needed. In our experience of approximately 100 adult pheochromocytomas and a small number of children, preoperative imaging signs of malignancy are rare and laparoscopic surgery is generally feasible even with larger tumours. Patients with locally advanced or metastatic disease at presentation should be treated by a combination of surgical debulking followed by adjuvant treatment to stabilise disease and treat symptoms.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


