Fig. 3.1
Chemical structure of hydrocortisone (cortisol) showing the four rings, A through D, along with numbering system for the carbon molecules. Also shown are the important features necessary for glucocorticoid activity (circled in red). These include hydroxyl group at positions 11, 17, and 21 as well as a keto group at position 3 and a double bond at 4–5 positions. It should be noted that the chemical structure of cortisone is identical to that of hydrocortisone with the exception that there is a keto group (=O) instead of a hydroxyl group (OH) at the 11 position
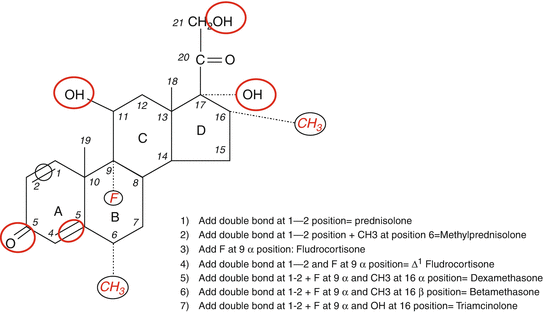
Fig. 3.2
Modifications in the basic chemical structure of cortisol can yield different compounds with variable anti-inflammatory and Na-retaining effects shown in red color and black circles. While some compounds have predominantly Na-retaining effects (e.g., fludrocortisone), others have potent anti-inflammatory effects without significant mineralocorticoid activity (e.g., dexamethasone). The figure shows specific addition to the basic structure resulting in the formation of commonly used synthetic glucocorticoids
It is important to point out that all synthetic glucocorticoids have alterations (double bond at 1–2 positions) or additions to positions 6, 9, and 16 (Fig. 3.2). A double bond at 1–2 positions of the hydrocortisone molecule results in the formation of prednisolone, and further addition of a methyl group at the 6 position results in the formation of methylprednisolone that possesses enhanced anti-inflammatory effects in comparison to the Na-retaining properties. The addition of the same (double bond at 1–2 positions) to cortisone results in the formation of prednisone. It is also important to point out that cortisone is not biologically active as it has a double bond at position 11 and would require the enzyme 11-β-hydroxysteroid dehydrogenase (11-β-HSD) to convert it to the active form, namely, hydrocortisone or cortisol [19, 25–27]. Likewise, prednisone has to be converted to the active moiety, prednisolone by the same enzyme [19, 25–27]. The addition of fluoride at position 9 to the hydrocortisone molecule results in the formation of fludrocortisone which has the highest mineralocorticoid activity among all synthetic adrenal steroids and is also a potent glucocorticoid as well [17]. In contrast, the addition of fluoride at position 9 and a double bond at 1–2 and a methyl group at position 16 results in the formation of dexamethasone with the most potent anti-inflammatory and the least (almost nonexistent) mineralocorticoid activity. Betamethasone (16-β-methyl group) has a similar structure to dexamethasone (16-α-methyl group) and is widely used as nasal and aerosol sprays for allergic and inflammatory lung disease. Alterations in the basic structure of the cortisol molecule near C-18 tend to decrease the mineralocorticoid effects of these compounds. The relative anti-inflammatory and Na-retaining effects of commonly used glucocorticoids [2, 3, 20, 21] are shown in Table 3.1.
Table 3.1
Commonly used oral and parenteral preparations of glucocorticoids
Dose equivalent in mgb | Anti-inflammatory potency | Na-retaining/mineralocorticoid potency | |
|---|---|---|---|
Hydrocortisone | 20 | 1 | 1.0 |
Cortisone acetatea | 25 | 0.8 | 0.8 |
Prednisonea | 5 | 4 | 0.8 |
Prednisolone | 4 | 4 | 0.8 |
Methylprednisolone | 4 | 5 | 0.5 |
Triamcinolone | 4 | 5 | Negligible |
Dexamethasone | 0.375–0.75 | 30 | Negligible |
Metabolism of Glucocorticoids
The major site of cortisol metabolism is the liver where it is reduced, oxidized, or hydroxylated and the products of these reactions are conjugated with sulfate or glucuronic acid and excreted in the urine [1, 3]. The main pathway in the metabolism of glucocorticoids is reduction of the 3-keto (by 3 α-HSD) and delta-4 double bond (by 5 α and 5 β reductase). The 11-β-HSD type 1 is a key enzyme in cortisol metabolism as it possesses dehydrogenase and reductase activities [25–27]. The enzyme has the ability to inactivate cortisol by oxidation in several tissues such as liver, bone, adipose tissue, and the central nervous system while the reverse (i.e., reduction or conversion of cortisone into cortisol) occurs primarily in the liver. Additional pathways of cortisol metabolism include oxidative removal of the side chain (to yield a 19-carbon molecule) or hydroxylation at position 6 to yield 6-β-hydroxycortisol. The latter pathway predominates when cortisol secretion is excessive [28, 29].
Several factors can alter hepatic metabolism of cortisol such as a change in thyroid hormone secretion where hyperthyroidism accelerates, while hypothyroidism decreases cortisol metabolism [30, 31]. A recent study showed that cortisol metabolism and clearance are decreased during critical illness that accounts, in part, for the continued rise in serum levels [32]. Renal failure does not influence enzymatic metabolism of cortisol although the clearance of the glucuronide-associated metabolites is diminished. No significant changes in the clearance of glucocorticoids are observed in patients on peritoneal dialysis. However, the clearance of synthetic glucocorticoids is increased in patients with renal failure on hemodialysis [33]. A recent study showed that moderate impairment in renal function (eGFR, 30–60 ml/min/1.73 m2) results in an increase in baseline AM serum cortisol level as well as a higher serum level following dexamethasone administration [34]. The latter finding should be taken into account when one interprets the results of dexamethasone suppression test.
The other site of cortisol metabolism is the kidney where cortisol is converted into cortisone by 11-β-HSD type-2 enzyme which prevents cortisol from binding to the mineralocorticoid receptor [26, 27]. Synthetic glucocorticoids are metabolized through similar pathways: reduction, oxidation, hydroxylation, and conjugation [20, 23, 24]. Their metabolism is also subjected to similar alteration to those noted with cortisol. Importantly, however intake of other drugs will have a more dramatic impact on the metabolism of most synthetic glucocorticoids. For example, mitotane [35], phenytoin [36, 37], carbamazepine [9] phenobarbital [38], and rifampin [28, 39] increase 6-β-hydroxylation and as such accelerate the metabolism of halogenated glucocorticoids including dexamethasone, betamethasone, and fludrocortisone. The clearance of glucocorticoids is also decreased in women on oral contraceptives [40, 41] and others on estrogen replacement therapy as a result of increased transcortin. Similar outcomes are observed in patients receiving strong inhibitors of CYP 3A4 such as clarithromycin [42].
Pharmacokinetics of Synthetic Glucocorticoids
In contrast to cortisol, most synthetic glucocorticoids except prednisolone [21–24] have a very low affinity for CBG. Prednisolone has approximately half of the affinity for CBG as compared to cortisol, while the rest of synthetic glucocorticoids have minimal (<1%) affinity to the binding protein. Other than cortisol or prednisolone, synthetic glucocorticoids in the circulation are either weakly bound to albumin (60–70%) or are in the free form (30–40%). The plasma half-lives of synthetic glucocorticoids are longer than that of cortisol, and there are significant interindividual variations that relate to genetic variation and the influence of simultaneously used drugs as discussed earlier. Clearance of synthetic glucocorticoids such as prednisone and prednisolone decreases with age and the time of drug administration being slower in the morning than later in the day. A small percentage of administered glucocorticoids can be detected in breast milk of nursing women.
The half-life of endogenously synthesized cortisol in the circulation is variable but ranges between 60 and 110 min when the concentration is within the maximal binding capacity of CBG which is 22–25 μg/dL [4]. When cortisol concentrations reach levels above that, the proportions of free and albumin-bound cortisol are increased; and thus, the free fraction is higher than usual 8–10%. The half-life of exogenously administered hydrocortisone is also variable depending primarily on the dose, presence of liver and kidney diseases, as well as other comorbidities such as critical illness [32]. The half-lives of synthetic glucocorticoids (other than cortisol and perhaps prednisolone) are higher than that of cortisol reaching nearly 3–4 h for dexamethasone. It is important to emphasize the fact that there are significant individual variability in the activity of enzymes involved in glucocorticoid metabolism and therefore the prospective half-life of each compounds in humans [23]. Furthermore, the concomitant intake of other drugs that alter the activity of enzymes involved in glucocorticoid metabolism can also modulate the plasma half-life of each glucocorticoid [9, 24, 35–39]. In addition, concurrent illnesses such as thyroid diseases [30, 31] or liver failure [43, 44] can modulate the effects of glucocorticoids and alter their plasma half-lives.
As discussed elsewhere, cortisol metabolism and clearance are decreased during critical illness which accounts, in part for the continued rise in serum levels [32]. It is not known whether this will also be true about other glucocorticoids. Also as noted elsewhere, the clearance of cortisol as well as that of other glucocorticoids is increased in patients with hyperthyroidism [30, 31] and in patients with renal failure during hemodialysis [33]. In light of the low binding of synthetic glucocorticoids to transcortin and albumin, patients with nephrotic syndrome do not experience major changes in synthetic glucocorticoid clearance as compared to those with normal binding proteins [22, 33]. Fluorinated glucocorticoids such as dexamethasone and betamethasone cross the placenta and therefore would impact fetal tissue and suppress the fetal adrenal cortex. In contrast, the placenta inactivates prednisone and prednisolone. Therefore, betamethasone would be the preferred glucocorticoid to use when the intended action is directed at the fetus such as enhanced fetal lung maturity in those where premature delivery is anticipated [45]. However, when glucocorticoids are used to treat a pregnant woman with a glucocorticoid-responsive illness, the preferred choice would be prednisone/prednisolone and not dexamethasone. The clearance of exogenously administered synthetic glucocorticoids is slowed in patients with advanced liver failure as a result of decreased metabolism.
As stated earlier, the pharmacokinetics of glucocorticoids can be modified by certain drugs that affect glucocorticoid metabolism. Other drugs that can influence glucocorticoid pharmacokinetics include those that reduce the GI absorption such as aluminum/magnesium hydroxide but not the proton pump inhibitors or the H-2 blockers [44, 46]. It is important to appreciate the fact that there are genetic differences in patients’ ability to metabolize drugs over and above the presence of associated illnesses or concurrent medications use. It is clear however that subjects who metabolize glucocorticoids slowly tend, as one would predict, to have a higher chance for adverse events [47].
Routes of Administration of Commonly Used Glucocorticoids
The rate of absorption of synthetic glucocorticoid through the GI tract is quite similar among available preparation. Glucocorticoids can be administered intravenously, intramuscularly, intra-articularly, and orally, as a nasal aerosol, topically on the skin, conjunctiva, or other mucous membranes such as the buccal and rectal mucosa [20, 48–51]. Glucocorticoids are generally insoluble in water, and thus they are formulated primarily as salts such as sodium phosphates or succinates for intravenous use. While hydrocortisone is absorbed quickly after an intramuscular injection, other glucocorticoids are absorbed at a much slower and at times unpredictable rate such as cortisone acetate and triamcinolone. Absorption after intra-articular injections occurs slowly over days to weeks and leads to continuous systemic exposure that is often associated with adverse effects including suppression of the HPA axis. The absorption after oral administration is rapid occurring within 30–45 min and can be decreased by concomitant use of calcium/magnesium-containing drugs. Most topical and inhaled glucocorticoids such as fluticasone are absorbed to a certain degree and are often associated with adverse events. Many patients on chronic inhaled glucocorticoids have partial suppression of the HPA axis. Absorption of topically administered glucocorticoids is variable and depends to a large extent on the drug used, the skin abnormality and its integrity where inflamed and open skin tends to result in much higher absorption as compared to an intact skin [49, 50]. A thin region of the skin (face as compared to the arms) and the presence of occlusive dressing can also increase absorption.
Glucocorticoid Actions
Although the molecular mechanisms of glucocorticoid actions are discussed in detail elsewhere, a brief description is included in this chapter. Glucocorticoids exert their effects on many cells and tissues throughout the body through binding to the glucocorticoid receptor (GR) that is a member of the nuclear hormone receptor superfamily. GR resides primarily in the cytoplasm and once activated it undergoes conformational changes through a process that involves dissociating from heat shock proteins. Subsequently, the ligand-bound GR complex translocate into the nucleus where it binds to hormone-responsive or glucocorticoid-responsive elements in the target cells to induce or suppress gene transcription [52–56]. It should not be surprising to note that the response to glucocorticoids in a particular cell will depend to a large degree on the specific glucocorticoid used as well as the number of hormone-responsive elements associated with that gene. This is consistent with the known variable effects of glucocorticoids on different cell types in the body. It is well known that glucocorticoid and mineralocorticoid receptors share considerable homology that allows activation of either receptor by any of the two hormones. However, the specificity of the mineralocorticoid receptor is maintained by the presence of the enzyme 11-β-hydroxysteroid dehydrogenase in renal epithelial cells which inactivates cortisol by converting it to cortisone and thus allowing only mineralocorticoids to bind to that receptor [25, 27].
The anti-inflammatory and sodium-retaining potencies of synthetic glucocorticoids are quantified in comparison to the naturally occurring molecule, cortisol, using bioassays that rely on the known effects of the hormone. For example, the anti-inflammatory effects can be measured by the ability of the drug in question to eliminate the inflammatory response to an oil injection in rats. An example of methods assessing mineralocorticoid effects includes the ability of the tested drug to lower sodium excretion in adrenalectomized rats. Other assays assess potency by evaluating the drop in eosinophil count with the administration of the drug to be tested. It is not clear that such an assay represent an accurate assessment of the ant-inflammatory potency of glucocorticoids. While the assays are useful, they are imperfect as they do not account for many other variables involved in the process, including the absorption, bioavailability, and individual differences.
Physiological and Supraphysiological Effects of Glucocorticoids
Glucocorticoids exert extensive physiological effects on many tissues throughout the body some of which are direct effects at the cellular level and others are indirect through effects on homeostatic mechanisms [12]. The fact that glucocorticoids are essential for life is best appreciated in patients with deficiency. Cortisol, the most potent endogenous glucocorticoid is one of the most important defense mechanism the body has in the early and late phases of major stresses including critical illness [2, 12, 57]. In that respect, glucocorticoids in general and cortisol in particular help maintain homeostasis through their permissive effects on catecholamine receptors in vascular smooth muscles as well as their Na-retaining properties [58–62]. In addition, this class of steroids functions as a source of energy by stimulating gluconeogenesis and facilitating the lipolytic effects of catecholamines. Glucocorticoids stimulate adipocyte differentiation and adipogenesis through activation of key enzymes such as lipoprotein lipase. Excess glucocortico id favors deposition of visceral rather than peripheral fat which explains the clinical picture of central obesity seen in such individuals. It is likely that the increase in visceral fat in patients with endogenous or exogenous glucocorticoid excess is due to the fact that the glucocorticoid receptor is highly expressed and also the type-1 isoenzyme of 11-β-hydroxysteroid dehydrogenase is highly expressed. The latter feature promotes the conversion of cortisol from cortisone.
The impact of excess glucocorticoids on other hormones such as growth hormone and gonadotropins is quite significant. Growth hormone secretion is suppressed with excess glucocorticoids and this becomes quite significant in children as stunted growth is commonly observed [26, 61]. Variable degrees of suppression of gonadotropin secretion are commonly observed leading to signs and symptoms of hypogonadism. This effect has deleterious consequences on bone health as it adds to the negative effects of excess glucocorticoids on the bone [26, 63]. The latter effects on the bone are due to inhibition of osteoblast function, inhibition of calcium absorption, and at the same time increased renal Ca excretion. That is why osteopenia or frank osteoporosis is commonly seen in patients with excess glucocorticoids. Excess glucocorticoids cause progressive insulin resistance that often leads to the development of frank diabetes mellitus. In contrast, deficiency of glucocorticoids leads to decreased gluconeogenesis that can lead to hypoglycemia.
Excess glucocorticoids cause catabolic changes in the skin leading to collagen breakdown, skin thinning, and easy bruisability. In addition to their effects on promoting insulin resistance in the muscle, excess glucocorticoids cause atrophy (but not necrosis) of specific fibers inducing a proximal myopathy that is one of the common manifestations in patients exposed to excessive glucocorticoids from endogenous or exogenous sources.
Related posts:
 Pheochromocytomas and Paragangliomas: Genetics and Pathophysiology
Pheochromocytomas and Paragangliomas: Genetics and Pathophysiology
 Adrenal Zonation and Development
Adrenal Zonation and Development
 Diagnosis and Management of Adrenal Insufficiency
Pheochromocytomas and Paragangliomas: Genetics and Pathophysiology
Diagnosis and Management of Adrenal Insufficiency
Pheochromocytomas and Paragangliomas: Genetics and Pathophysiology
 Adrenal Cushing’s Syndrome: Updates on Overt and Mild Hypercortisolism
Adrenal Cushing’s Syndrome: Updates on Overt and Mild Hypercortisolism
 Diagnosis and Management of Adrenal Insufficiency
Diagnosis and Management of Adrenal Insufficiency
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


