Reactivation of fetal hemoglobin (HbF) expression is an important therapeutic option in adult patients with hemoglobin disorders. The understanding of the developmental regulation of γ-globin gene expression was followed by the identification of a number of chemical compounds that can reactivate HbF synthesis in vitro and in vivo in patients with hemoglobin disorders. These HbF inducers can be grouped in several classes based on their mechanisms of action. This article focuses on pharmacologic agents that were tested in humans and discusses current knowledge about the mechanisms by which they induce HbF.
Human hemoglobin is a tetrameric molecule comprised of two pairs of identical polypeptide subunits, each pair encoded by a different family of genes. The human α-like globin genes (ζ, α1, and α2) are located on chromosome 16 and the β-like globin genes (ε, G γ, A γ, δ, and β) are located on chromosome 11. During development, sequential switches take place in both the α- and β-like globin clusters that result in the production of six different types of hemoglobins. Synthesis of hemoglobin begins during the first weeks of embryonic development in the yolk sac where three different hemoglobins are made: (1) Hb Gower 1 (ζ2ε2); (2) Hb Portland (ζ2γ2); and (3) Hb Gower 2 (α2ε2). By the 13th week of gestation, the site of erythropoiesis shifts from the embryonic yolk sac to the fetal liver where fetal hemoglobin (HbF; α2γ2) starts to accumulate as the embryonic ε-globin gene is turned off. Shortly before birth, a second switch occurs as the site of erythropoiesis shifts from the fetal liver to the bone marrow. As a result of this second switch, the two adult hemoglobins (HbA, α2β2; and HbA 2 , α2δ2) gradually replace HbF. By the end of the first year of life, the hemoglobin composition is approximately 97.5% HbA, 2% HbA 2 , and 0.5% HbF.
In adult life, the residual amounts of HbF are distributed unevenly among red cells. HbF is concentrated in a subpopulation of erythrocytes known as “F cells.” The amount of HbF and the number of F cells are genetically determined and vary within a relatively narrow range in normal individuals. Such factors as acute blood loss, pregnancy, and bone marrow transplantation that lead to erythropoietic stress can result in an increase in baseline levels of HbF. In addition, inheritance of a variety of genetic determinants, such as sickle cell disease (SCD), thalassemia, and hereditary persistence of HbF, can also lead to higher levels of HbF and F cells in adult life.
The levels of HbF in erythrocytes account for a large part of the clinical heterogeneity observed in patients with SCD and β-thalassemia. Patients with SCD from certain regions of Saudi Arabia and India have an unusually mild clinical sickling disorder associated with high levels of HbF. Moreover, the large multicenter study of the natural history of SCD (Cooperative Study of SCD) identified HbF as a major predictor of several complications, including painful events, acute chest syndrome, and mortality. In addition, patients with SCD or β-thalassemia who co-inherit a genetic determinant for hereditary persistence of HbF usually have a mild clinical disorder. These clinical and epidemiologic observations provided important clues about the beneficial role of HbF in modulating the pathophysiology of these disorders. In SCD, a high level of HbF interferes with the polymerization of HbS and prevents sickling of red blood cells. On the other hand, in β-thalassemia, a high level of γ-globin chain synthesis decreases non-α:α chain imbalance and ameliorates the anemia. Based on all these observations, it was proposed that pharmacologic induction of HbF production might be an effective therapeutic strategy for ameliorating the severity of SCD and β-thalassemia.
Regulation of γ-globin gene expression
Gene transcription of the γ-globin genes, and of other globin genes present in the β-globin cluster, is controlled by complex molecular mechanisms involving cis -acting elements, represented by specific nucleotide sequences, such as the cluster control region (LCR) and the promoters of the different globin genes and trans -acting elements, such as transcription factors and chromatin remodeling proteins.
The LCR consists of at least five DNase I hypersensitive sites (HS1–HS5). Each HS contains one or more binding motifs for three erythroid-specific transcription factors (GATA-1, NF-E2, and EKLF ) in addition to binding sites for ubiquitous DNA binding proteins. HS1 to HS4 are formed only in erythroid cells. The transcriptional-enhancer activity of the LCR resides mostly in HS2 and HS3. HS3 is believed to be involved in γ-globin activation during fetal-stage development and in β-globin activation during adult life. It is postulated that EKLF bound to HS3 may provide a competitive advantage for the interaction of the LCR with the β-globin promoter over its interactions with the γ-globin promoter, facilitating hemoglobin switching after birth. The role of the LCR in switching has been conceptualized by the competition model based on the presence of developmental stage-specific transcription factors that mediate its interactions with the individual globin gene promoters. However, significant uncertainty exists regarding the potential effects of the LCR on chromatin conformation in the β-globin gene cluster. In patients with Hispanic δβ-thalassemia in which the LCR is deleted, the β-globin cluster chromatin domain is in a closed, DNase I-resistant, transcriptionally inactive conformation, suggesting that the LCR functions to open chromatin in addition to its direct role in globin gene activation. However, when the β-globin LCR was deleted from the endogenous mouse β-globin cluster in embryonic stem cells and somatic cell lines, β-like globin transcript levels were reduced, whereas the switching pattern during development remained normal and the chromatin of the β-globin gene cluster existed in an open DNase I–sensitive conformation. These studies suggest that the LCR is not necessary for the establishment of an open chromatin cluster and that its primary function is that of an enhancer for transcriptional activation of the globin genes. Furthermore, these studies also suggested that the sequences conferring developmental stage-specific expression reside in the regions immediately flanking the globin genes. These findings are consistent with previous studies by Arcasoy and coworkers, who found autonomous silencing of the γ-globin genes in adult transgenic mice despite the absence of the LCR.
GATA-1, EKLF, and NF-E2 are the best-characterized tissue-specific transcription factors involved in the regulated expression of the β-like globin genes. All these factors bind to DNA elements in the β-like globin genes. GATA-1 and EKLF are essential for active chromatin hub formation. Contacts between the LCR and the β-like globin genes do not form in their absence. In contrast, NF-E2 seems to be dispensable for β-globin active chromatin hub formation. The essential role of GATA-1 in inducing β-globin LCR chromatin structure remodeling is also supported by recent studies of enforced GATA-1 expression in nonerythroid cells. Transcription factors that activate the γ-globin gene promoter include the FKLF2, NF-E4, and SSP. Other studies have provided evidence that long-range interactions between the LCR and the active β-globin genes requires the binding to the LCR of a multimeric transcriptional complex formed by the nuclear protein Nuclear LIM interactor and its erythroid binding partners GATA-1, TAL-1, and LMO2.
Pharmacologic Reactivation of HbF Synthesis
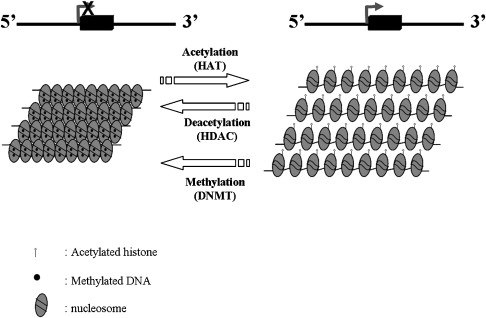
During the last two decades, a great deal of effort has been focused on the pharmacologic induction of HbF in patients with hemoglobin disorders. Multiple drugs including 5-azacytidine, hydroxyurea, erythropoietin, and butyrate were found to have in vivo activity in patients with these disorders. Histone deacetalyse (HDAC) inhibitors, such as butyrate, and related compounds and DNA methylation inhibitors, such as 5-azacytidine and decitabine, are believed to exert their effects through epigenetic modifications in the β-globin gene cluster ( Fig. 1 ). Despite the controversy over the exact mechanism of induction of HbF by 5-azacytidine and hydroxyurea, considerable progress has been made in the development of these agents as drugs that can induce HbF in patients with hemoglobin disorder.
DNA Methyltransferase Inhibitors
DNA methylation is a mechanism of gene silencing in which methyl groups become covalently attached to the 5-carbon position of a cytosine residue at a “CpG” site. This epigenetic process occurs during S phase and is catalyzed by the DNA methyltransferase family of enzymes. Genes with heavily methylated promoter regions cannot be transcribed and are effectively silenced (see Fig. 1 ).
Several studies that were published in the early 1980s showed a strong inverse correlation between γ-globin gene expression and DNA methylation at the γ-globin gene promoters. During the embryonic and adult stages of development, methylation of the γ-globin promoters was shown to correlate with silencing of the γ-globin genes. In contrast, hypomethylation of the γ-globin gene promoters during the fetal stage was shown to correlate with expression of the γ-globin genes. Similarly, the inactive β-globin gene was shown to be methylated in fetal life, whereas the active β-globin gene was shown to be hypomethylated in adult life. These observations support a model in which differences in the methylation pattern of the γ-globin gene in differentiating erythroblasts at different stages of development may be the result of fetal stage-specific demethylation associated with transcription activation. Therefore, considerable efforts have been focused on the use of DNA hypomethylating agents for the pharmacologic stimulation of HbF production in adult life.
5-Azacytidine was the first prototype of an agent that induces HbF by targeting epigenetic gene silencing. This drug was first used in the United States during the 1970s as an antileukemic agent and later in the 1980s as an HbF inducer in patients with hemoglobin disorders. 5-Azacytidine was recently approved by the Food and Drug Administration for the treatment of patients with myelodysplastic syndrome, although the mechanisms responsible for its beneficial effects in this disorder remain unknown. The ability of 5-azacytidine to stimulate HbF production was first demonstrated in the anemic baboon where HbF levels increased up to 70% to 80% of total hemoglobin after treatment with this drug. These observations provided strong support for the hypothesis that γ-globin gene expression could be pharmacologically induced in vivo by DNA hypomethylation at the promoters of the γ-globin genes, resulting in increased levels of HbF. These encouraging data led to clinical trials of 5-azacytidine in a small number of patients with SCD and β-thalassemia. This treatment resulted in significant increases in HbF levels (7%–23%); F cells (11%–50%); and total hemoglobin (1–4 g/dL). The therapeutic effects of this drug were associated with a decrease in the percentage of dense cells in patients with SCD and of the non-α:α chain imbalance and transfusion requirements in patients with β-thalassemia. Despite these promising results, clinical trials with this agent were not continued because of concerns over potential carcinogenic effects of 5-azacytidine, which was previously shown to increase the incidence of tumors in an animal model.
The more recent introduction of decitabine as a DNA hypomethylating agent resulted in renewed interest in the use of DNA hypomethylation for the induction of HbF ( Fig. 2 ). Three clinical trials have been reported by the same group in which decitabine was administrated to patients with SCD. In the first two studies, the drug was administrated to a total of 15 patients by intravenous injections at doses of 0.15 to 0.3 mg/kg/day. In the third study, decitabine was administrated to eight patients by subcutaneous injections at doses similar to those used in the previous studies. This therapy resulted in significant increases in mean γ-globin synthesis, HbF levels, and F cells by 10%, 13%, and 33%, respectively. Interestingly, 100% of patients with SCD showed an increase in HbF production in response to decitabine, including several patients who had previously failed to respond to hydroxyurea. With decitabine treatment, total hemoglobin levels increased by 2 g/dL, whereas the reticulocyte count decreased, suggesting a decrease in hemolysis. The increase in the levels of HbF was associated with significant improvement in several parameters that are important in the pathophysiology of vasoocclusion, such as red blood cell adhesion, endothelial damage, and activation of the coagulation pathway. The authors also demonstrated a decrease in DNA methylation at the promoters of the γ-globin genes. The only toxicity that was observed in these studies was transient neutropenia. Interestingly, the platelet count and the proportion of megakaryocytes and erythroid cells in the marrow increased, without a corresponding decrease in marrow cellularity. The significance of these observations is not yet clear.
Despite these encouraging short-term studies, the clinical effectiveness and the potential long-term toxicities of decitabine treatment are not known at this stage. An early study reported that this agent was not carcinogenic in the rat model. Other relatively short-term studies by Yang and colleagues in patients with leukemia treated with decitabine did not show an increase in incidence of secondary tumors at 2 to 5 years after initiation of therapy. More recent studies have suggested that treatment of mice with a genetic disposition for colon or lung cancer with decitabine results in a marked reduction in tumor formation. It is speculated that this reduction in tumorogenesis may be a reflection of the prevention of methylation of tumor-suppressor genes, a process that is believed to be important in the pathogenesis of some cancers. These studies suggested that decitabine may have some potential in the chemoprevention of cancer. Larger and longer-term studies are needed to confirm the safety and efficacy of decitabine in patients with hemoglobin disorders.
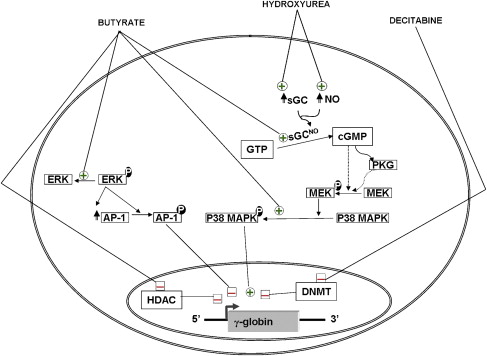
The molecular mechanisms of induction of HbF by 5-azacytidine and decitabine are not fully understood. These agents were first introduced based on their ability to inhibit methylation of newly synthesized DNA within the promoters of the γ-globin genes. One of the five CpGs within the promoters of the fetal γ-globin genes that could be hypomethylated on exposure to these agents coincides with a DNA sequence that is occupied by proteins that participate in the switch from fetal to adult globin expression. Methylation of this CpG favors the binding of a repressor, which might be responsible in part for γ-globin repression in adult erythroid cells. In contrast, hypomethylation of this same CpG can enhance the binding of an activator of γ-globin gene expression. In addition, recent experiments in baboons showed that DNA hypomethylation induced by treatment with decitabine is associated with histone hyperacetylation at the γ-globin promoters. Taken together, these data suggest that DNA methylation may result in changes in chromatin structure that enhance the binding of transcription factors, leading to derepression and transcriptional activation of γ-globin gene expression in adult life (see Fig. 2 ).
Despite these data, the causal role of DNA hypomethylation in the induction of HbF expression by these analogs remains controversial. It is well known that a partial switch from adult to fetal hemoglobin production takes place during accelerated erythropoiesis, as seen following acute blood loss. This results in augmented F-reticulocytosis and a corresponding increase in HbF levels. Thus, some investigators proposed that 5-azacytidine treatment may result in injury to bone marrow progenitors, leading to accelerated erythropoiesis and an increase in HbF levels. In support of this hypothesis, hydroxyurea, another S-phase–specific chemotherapeutic agent, was shown to increase HbF levels in anemic baboons. Because hydroxyurea does not have a direct effect on DNA methylation, it was proposed that the induction of HbF by 5-azacytidine is unlikely to be a result of its DNA hypomethylating activity. Moreover, molecular examination of DNA methylation in bone marrow cells from a patient who failed to respond to 5-azacytidine with increased γ-globin synthesis revealed hypomethylation of the γ-globin promoters. Thus, the exact mechanism of induction of HbF by hypomethylating agents remains shrouded in controversy.
Histone Deacetylase Inhibitors
The acetylation of histones and nonhistone proteins at the ε-amino group of lysine residues is a reversible posttranslational process. Acetylation of histones is commonly associated with transcriptional activation of genes and is thought to be responsible both for a local “open chromatin” structure required for promoter activation and for impacting the binding of multiple transcription factors directly. Histone acetylation can be achieved either by acetyltransferases alone or by adenosine triphosphate–dependent nucleosome remodeling complexes, such as SWI-SNF complexes. In contrast, removal of acetyl groups by HDACs frequently accompanies suppression of gene activity, and HDACs are often found as components of transcriptional repressor complexes. Different signaling events may switch the association patterns from acetyltransferases to deacetylases, or vice versa, providing a potential mechanism for dynamic control of acetylation in vivo. The comparative analysis of the histone acetylation patterns in the human β-globin cluster between the fetal and adult stages of development showed that the LCR was acetylated at the same level in both fetal and adult erythroblasts, whereas the acetylation in the globin promoter regions correlated with the state of transcription.
Several lines of experimental evidence have suggested that HDAC inhibition results in increased HbF synthesis. In vitro studies performed in erythroleukemia cell lines and in primary erythroid cultures have shown that several HDAC inhibitors, such as TSA, MS-275, scriptaid, and apicidin, simulate HbF synthesis. In vivo, butyrate, a natural short-chain fatty acid and a well-known HDAC inhibitor, was shown to increase embryonic globin gene expression in chicken adult erythroid cells pretreated with 5-azacytidine. Furthermore, infants of diabetic mothers were shown to have a delay in γ- to β-globin switch after birth. This was attributed to the elevated plasma α-amino butyric acid levels that result from ketosis during pregancy. Moreover, exposure to butyrate was shown to result in increase HbF synthesis in adult baboons and the fetal to adult globin gene switch was significantly delayed in butyrate-treated lamb fetuses.
In 1993, arginine butyrate was administrated to six patients, three with SCD and three with β-thalassemia. This initial study provided evidence that arginine butyrate infusion can increase HbF production in vivo in patients with hemoglobin disorders. However, a subsequent study showed that the continuous infusion of arginine butyrate did not result in a sustained HbF production and did not improve the hematologic parameters in patients with SCD or β-thalassemia. However, when arginine butyrate was administered intermittently at lower doses in a subsequent study in patients with SCD, this resulted in a sustained induction of HbF production in most patients. The success of this regimen was attributed to its ability to avoid toxicity to erythroid precursors that might result from continuous exposure to high levels of butyrate. However, not all patients with SCD responded to butyrate treatment: interestingly, all patients whose baseline HbF levels were greater than 2% responded to treatment, whereas all patients with HbF levels less than 2% were resistant to this treatment. Although pulse administration of butyrate can result in a significant improvement in the anemia in patients with β-thalassemia, most treated patients continue to suffer from profound anemia.
Given the difficulty of administration of large volumes of arginine butyrate through central venous catheters, orally administered butyrate compounds have been tested to treat patients with SCD and β-thalassemia. Both sodium phenylbutyrate and isobutyramide resulted in some stimulation of HbF synthesis. However, the induction of HbF with these agents was modest and patient compliance was a significant challenge. As a result, clinical development of these agents did not go forward.
It is widely believed that butyrate increases γ-globin gene expression by increasing histone acetylation at their promoters. Recently, using primary erythroid cultures derived from peripheral blood of normal donors and patients with SCD, the authors analyzed the epigenetic changes induced by butyrate at the promoters of different genes of the β-globin cluster. This analysis showed that butyrate exposure results in a true reversal of the normal developmental switch from γ- to β-globin expression. This is associated with increased histone acetylation and decreased DNA methylation of the γ-globin genes, with opposite changes at the promoter of the β-globin gene. In vivo foot-printing studies performed in erythroblasts of patients who responded to butyrate by increasing HbF production revealed alterations in DNA binding at the proximal region of the γ-globin gene promoter. Butyrate was also shown to activate signaling through the activation of soluble guanylate cyclase (sGC) and p38 MAP kinase pathways and inhibition of ERK MAP kinase pathway (see Fig. 2 ). However, it is not yet clear how these signaling activities lead to the transcriptional activation of the γ-globin genes. More recently, butyrate was also shown to increase HbF production in patients with SCD by increasing the efficiency of translation of γ-globin mRNA.
Related posts:
 HbE/β-Thalassemia: Basis of Marked Clinical Diversity
HbE/β-Thalassemia: Basis of Marked Clinical Diversity
 Allogeneic Cellular Gene Therapy for Hemoglobinopathies
Allogeneic Cellular Gene Therapy for Hemoglobinopathies
 Iron Overload in Thal assemia and Related Conditions: Therapeutic Goals and Assessment of Response to Chelation Therapies
Iron Overload in Thal assemia and Related Conditions: Therapeutic Goals and Assessment of Response to Chelation Therapies
 Protein Quality Control During Erythropoiesis and Hemoglobin Synthesis
Protein Quality Control During Erythropoiesis and Hemoglobin Synthesis
 Hemoglobin Gene Therapy for β-Thalassemia
Mouse Models of Inherited Cancer Syndromes
Hemoglobin Gene Therapy for β-Thalassemia
Mouse Models of Inherited Cancer Syndromes
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


