Fig. 10.1
The ominous octet. Adapted from DeFronzo R
The imbalance between alpha and beta cells is a novel mechanism implicated in the pathogenesis of Type 2 DM described in the last few years. It is due to a defective action of incretin hormones, mainly glucose-dependent insulinotropic polypeptide (GIP) and glucagon-like peptide 1 (GLP-1). This impaired action causes the failure to suppress glucagon release after a meal ingestion, increasing glucose concentration. The decrease in GPL-1 action in the brain together with insulin resistance (IR) in the central nervous system produces the dysregulation of food-intake behavior (less satiety). GIP and GLP-1 are released predominantly from L and K cells (in the proximal and distal intestine) following nutrient ingestion promoting insulin secretion, glucagon release inhibition, delaying gastric emptying, and enhancing satiety (controlling weight) with GLP-1 being much more effective than GIP. GLP-1 and GIP have multiple actions that enhance beta-cell response in a glucose-dependent fashion. In T2DM, the incretin response is diminished (Van Genugten et al. 2013).
Finally, another interesting mechanism described recently is the diminished action of the sodium-glucose cotransporter 2 (SGLT2) in the kidney. This defect decreases glucose excretion. The kidney contributes to glucose homeostasis by filtering and reabsorbing glucose back into the circulatory system. In non-diabetics, approximately 180 g of glucose is freely filtered per day, which is all reabsorbed in the proximal convoluted tubule. Two transporter families are involved with glucose reabsorption: glucose transporters (GLUTs) as passive transporters and SLGTs as secondary active transporters. Two of the six SLGTs are well characterized: (1) SLGT-1—with low capacity, located in small intestine and in the renal proximal convoluted tubule responsible for only 10 % of glucose re-absorption and (2) SLGT-2—with a high capacity of absorption, in the early proximal convoluted tubule, responsible for the remaining 90 % of glucose reabsorption (Vasilakou et al. 2013) (Fig. 10.1).
A better knowledge of the pathophysiology of T2DM and its comorbidities has challenged the development of many new drugs to safely treat hyperglycemia (Fig. 10.2) with the aim to reduce and maintain glucose concentrations as close to normal for as long as possible after diagnosis, preventing the development of chronic complications.
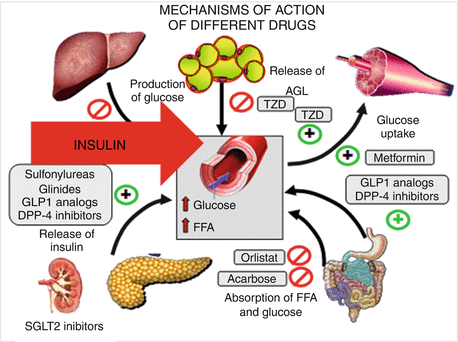
Fig. 10.2
Mechanism of action of different drugs for the treatment of type 2 diabetes
10.2 From the Classical Approach to the New Drugs in the Treatment of Type 2 Diabetes
10.2.1 Metformin
According to national and international guidelines, metformin is the recommended first-line oral therapy for the treatment of type 2 diabetes together with lifestyle intervention at the time of diagnosis. However, it could also be combined with almost all the rest of antidiabetic agents with the progression of the disease (Nathan et al. 2009).
10.2.1.1 Mechanisms of Action
Metformin is effective only in the presence of insulin, decreasing hepatic glucose output and increasing insulin-mediated glucose utilization in peripheral tissues (such as muscle and liver), particularly after meals. It has an antilipolytic effect lowering serum-free fatty acid concentrations reducing substrate availability for gluconeogenesis (UKPDS 1995).
Metformin ameliorates hyperglycemia by reducing insulin resistance. These effects have been linked to the expression of insulin receptors as well as tyrosine kinase activity along with the modulation of the incretin axis. Metformin also increases plasma GLP-1 levels and gene expression of the islet incretin receptor via peroxisome proliferator-activated receptor-α (PPAR-α). Whatever be the role, metformin through all these mechanisms primarily inhibits hepatic glucose output and significantly increases insulin sensitivity (Viollet et al. 2012).
The pharmacokinetics of metformin is described in Table 10.1.
Table 10.1
Pharmacokinetics of metformin
Bioavailability | 50–60 % absorption in the small intestine; estimated maximum concentration 0.9–2.6 h and 4–8 h with standard formulations with delayed absorption |
Plasmatic concentration | Maximum 1–2 μg/ml within 1–2 h after oral doses of 500–1,000 mg of the standard formulation; approximately 20 % less with the extended release |
Half-life | 6 h of removal |
Metabolism | Not metabolized |
Distribution | Concentration in most tissues similar to plasma; higher in the liver and kidney; the highest concentration in salivary glands and intestinal wall |
10.2.1.2 Doses, Associations, and Efficacy
Metformin should be indicated as monotherapy for the treatment of hyperglycemia in patients with type 2 diabetes who do not achieve the expected levels of glycemic control with a non-pharmacological approach based on diet, exercise, and diabetes education.
Metformin should be combined with sulfonylureas, glinides, alpha-glucosidase inhibitors, thiazolidinediones, GLP-1 agonists/analogs (exenatide, liraglutide), dipeptidyl peptidase-4 inhibitors (DPP-4), and insulin.
Metformin lowers HbA1c by 1.5%, similar to that achieved with sulfonylurea (Hermann et al. 1994). Metformin shows potential beneficial effects associated with the decrease of insulin resistance (Table 10.2).
Table 10.2
Effects of metformin against insulin resistance syndrome
Features of the insulin resistance syndrome | Effect of metformin on the features of the insulin resistance syndrome |
|---|---|
Hyperinsulinemia | Reduces hyperinsulinemia during fasting periods |
Abdominal obesity | Generally stabilizes weight, reduces weight gain, and facilitate weight loss |
Glucose intolerance or type 2 diabetes | Counteracts insulin resistance (increases the suppressive effect of insulin on hepatic glucose production and increases its muscle uptake). Slows the progression of prediabetes to diabetes, improves glycemic control in type 2 diabetes |
Dyslipidemia (↑ c-VLDL-TG, ↑ c-LDL, ↓ c-HDL) | Improves slightly the lipid profile |
Hypertension | No significant effect on blood pressure in most studies |
Procoagulant state | Some antithrombotic activity (e.g., Decreases PAI-1, fibrinogen, and platelet aggregation) |
Atherosclerosis | Facts of antiatherogenic activity in preclinical studies |
Initial doses (500 mg once a day) must be slowly increased at weekly intervals and administered two or three times a day before or together with meals considering a maximum dose of 2,550 mg/day.
10.2.1.3 Side Effects and Contraindications
At the start of the treatment, patients could present gastrointestinal side effects (abdominal discomfort, diarrhea, nausea, anorexia, and metallic taste). They can be minimized by titrating the dose gradually and take the tablets with meals.
Metformin reduces intestinal absorption of vitamin B12 in up to 30 % of patients, and lowers serum vitamin B12 concentrations in 5–10 %, but rarely causes megaloblastic anemia.
Metformin is weight neutral and does not produce hypoglycemia compared with other traditional oral agents (Bailey and Turner 1996).
Metformin should be used carefully in patients with impaired renal function (creatinine ≥1.5 mg/dl in men or ≥1.4 mg/dl in women and/or glomerular filtration rate <60 ml/m/1.73 m2). However, the last international guideline allows its use in patients with a glomerular filtration rate over 30–45 ml/m if they have received it before. In elderly patients, it is contraindicated if the estimated glomerular filtration rate is less than 60 ml/min/1.73 m2. Temporary suspension of metformin is mandatory 48 h prior to any intravascular contrast procedure.
Hypoxia and hypoperfusion states are considered a contraindication to metformin administration, in particular, acute heart failure, severe respiratory failure, and sepsis. The presence of severe liver disease and a history of lactic acidosis, alcohol abuse, or other functional disorders that could produce abnormal lactate metabolism are also considered as contraindications. At present, metformin is not recommended during pregnancy and lactation. Metformin is not effective as a primary therapy in type 1 diabetes.
Lactic acidosis remains as one of the main concerns because of the high case fatality rate. However, most cases occurred in patients with a shock state and/or tissue hypoxia or in the presence of several other predisposing conditions like impaired renal function and heart failure (Table 10.3).
Table 10.3
Clinical use of metformin
Alone or in combination with other oral antidiabetic agents and/or insulin in patients with type 2 diabetes inadequately controlled with diet and exercise |
Directions for use: available presentations—500, 850, and 1,000 mg. Simple or extended release. Better to administer with meals, and increase doses in a gradual way Maximum dose 2,550 mg/day (2,000 g/day in children) |
Contraindications: renal and hepatic impairment; heart or respiratory failure; hypoxemia; severe infection; alcohol abuse; history of lactic acidosis; pregnancy; temporary suspension during scans with intravenous contrasts |
Gastrointestinal adverse effects relieved by reducing doses; may interfere with the absorption of vitamin B12 and folic acid |
Risk: lactic acidosis in patients with contraindication; hypoglycemia in combination with other drugs or in moderate alcohol consumption |
10.2.2 Sulfonylureas
Sulfonylureas have been established in the treatment of Type 2 DM and were the first oral glucose-lowering medications to be introduced into clinical practice. Since then many different molecules had been developed (1st and 2nd generations of sulfonylureas)
10.2.2.1 Mechanisms of Action
Their main mechanism of action is the insulin release from pancreatic islet beta cells increasing insulin disposal during fasting and postprandial periods. There were described many other actions of sulfonylureas: Decrease of the exaggerated hepatic glucose overproduction and the improvement of insulin sensitivity in muscle and adipose tissue. The main source of sulfonylurea receptors is not only located in beta cells but also distributed in many organ tissues like myocardial cells.
The sulfonylurea receptor is a component of the ATP-dependent potassium channel (Aguilar-Bryan et al. 1995).
Sulfonylurea binding to its receptors leads to the inhibition of these channels, which alters the resting potential of the cell, leading to calcium influx and stimulation of insulin secretion in the beta cells and also to the blockade of ischemic preconditioning in myocardial cells. The net effect is an increased responsiveness of beta cells to both glucose and non-glucose secretagogues (such as amino acids), resulting in more insulin secretion at different blood glucose concentrations. Thus, sulfonylureas are useful only in patients with some remaining beta cell function (Ashcroft 1996).
The 1st generation sulfonylureas (tolbutamide, chorpropamide) have been stopped and there should be very few patients using them. The 2nd generation sulfonylureas, glipizide, glyburide (glibenclamide), gliclazide, and glimepiride, have structural characteristics that allow them to be given in much lower doses than the 1st generation. Nevertheless, they are equally effective in lowering blood glucose concentrations in spite of differences in absorption and metabolism, as well as in effective dose
10.2.2.2 Doses, Associations, and Efficacy
Sulfonylureas usually lower HbA1c by 1–2 %. They are most likely to be effective in patients whose weight is normal or slightly increased (Bressler and Johnson 1997).
They can be combined with any other drugs like metformin, TZD, incretin-mimetics, DPP4I, and/or insulin. Doses and pharmacokinetics of the SU are described in Table 10.4.
Table 10.4
Dosage and administration of sulfonylureas and meglitinides
Drug | Dose (mg/day) | Maximum effect (h) | Half-life (h) | Metabolites | Excretion |
|---|---|---|---|---|---|
Sulfonylureas | |||||
Tolbutamide | 500–3,000 | 3–4 | 4.5–6.5 | Inactive | Kidney |
Clorpropamide | 100–500 | 2–4 | 36 | Active or unmodified | Kidney |
Tolazolamide | 100–1,000 | 3–4 | 7 | Inactive | Kidney |
Glipizide | 2.5–40 | 1–3 | 2–4 | Inactive | Kidney 80 %, stools 20 % |
Glipizide LA | 5–20 | Constant/days | – | Inactive | Kidney 50 % stools 50 % |
Glibenclamide | 1.25–20 | 2–4 | 10 | Inactive and weakly active | Kidney 60 % stools 40 % |
Glibenclamide (micronized) | 1.5–12 | 2–3 | –4 | Inactive and weakly active | |
Glimepiride | 1–8 | 2–3 | 9 | ||
Glinides | |||||
Repaglinide | 0.5–4 each meal | 1 | 1 | Inactive | Stools |
Nateglinide | 60–120 each meal | 1,8 | 1.4 | Weakly active | Kidney 80 % Stools 20 % |
10.2.2.3 Side Effects and Contraindications
Sulfonylurea is usually well tolerated. Hypoglycemia and weight gain are the most common side effects (Gangji et al. 2007).
Hypoglycemia is associated with high SU doses, alcohol abuse, impaired renal function, co-administration of salicylates, sulfonamides, fibric acid derivatives (gemfibrozil), and warfarin, or after missing meals and/or exercising.
Weight gain has been described during the UKPDS trial in the SU’s cohort. A change in body weight, pooling the results of many publications of this trial, gave a mean increase in weight of 2.31 kg (95 % CI 1.31–3.32) in the sulfonylurea-treated groups compared with comparator groups. For this reason, SUs should not be the first-line drug election for obese Type 2DM patients.
It has been described unfavorable cardiovascular effects probably related to the blockade of ischemic preconditioning mentioned before, an endogenous mechanism that protects the heart from ischemic injury associated with hypoglycemic episodes producing electrocardiographic abnormalities and arrhythmias (Marques et al. 1997). Gliclazide showed less hypoglycemia and less association with CV mortality.
The three main problems in relation to treatment with insulin secretagogues are
Hypoglycemia
Weight gain
Cardiovascular risk (an open discussion to date)
Contraindications for treatment with insulin secretagogues are as follows: Type 1 diabetes, pregnancy, major surgeries, infections, severe trauma, allergy predisposition, and severe hypoglycemia risks (Table 10.5).
Table 10.5
Contraindications for the use of insulin secretagogues
Diabetes type 1 or pancreatic diabetes |
Pregnancy |
Major surgeries |
Infections or severe traumas |
History of sulfonylurea or a related molecules’ severe adverse reaction. e.g., sulfonamide (doesn’t exclude repaglinide) |
Predisposition to severe hypoglycemia (e.g., patients with significant hepatic or renal disease) |
The response to sulfonylurea can decrease over time. The more common factors are listed in Table 10.6.
Table 10.6
The most frequent causes of secondary failure of sulfonylurea
Patient-related factors |
Weight gain or inadequate diet |
Poor adherence to treatment guides |
Physical inactivity |
Stress |
Intercurrent diseases |
Comorbidities |
Disease-related factors |
Functional loss of pancreatic beta cells |
Increased insulin resistance |
Treatment-related factors |
Inadequate posology |
Concomitant diabetogenic drugs |
10.2.3 Meglitinides
10.2.3.1 Mechanism of Action
The meglitinides are short-acting glucose-lowering drugs. They are structurally different from sulfonylureas and exert their effects via different receptors in the beta cell surface, but activating the same insulin release pathway generating short and increased prandial insulin release because of their shorter half-life (Fuhlendorff et al. 1998).
10.2.3.2 Doses, Associations, and Efficacy
Doses have been described in Table 10.4
They can be administered alone as monotherapy or in combination with almost all the other antidiabetic drugs except sulfonylureas and rapid-acting insulin.
The clinical efficacy of meglitinide as monotherapy is almost similar to that of the sulfonylureas (Wolffenbuttel and Landgraf 1999).
10.2.3.3 Side Effects and Contraindications
Nateglinide is metabolized in the liver with renal excretion of active metabolites. In the presence of decreased renal function, the accumulation of these metabolites increases the risk of hypoglycemia. This drug must therefore be used cautiously in people with high risks of hypoglycemia.
Repaglinide is mainly metabolized by the liver with less than 10 % renal excretion. Dose adjustments with this agent do not appear to be necessary in patients with renal impairment and should be administered in patients with a glomerular filtration rate of up to 45 mg/dl.
There are no long-term studies of meglitinides to assess cardiovascular outcomes or mortality.
The advantages of repaglinide (the most popular meglitinide) over SUs are the ability to produce less hypoglycemia and the potential use in patients with moderate impaired renal function.
The most common side effects are hypoglycemia and a very slight increase of weight, but significantly lower than SUs.
10.2.4 Thiazolidinediones
Thiazolidinediones are antihyperglycemic drugs whose main action is to increase peripheral insulin sensitivity.
The 1st thiazolidinedione drug generation, troglitazone, approved in 1997, had to be retired because of liver toxicity. Rosiglitazone (a 2nd generation thiazolidinedione) has been recently associated with cardiovascular complications, and it is being used only in a few countries with severe restrictions. The 2nd generation thiazolidinedione in use without restriction is pioglitazone.
10.2.4.1 Mechanism of Action
Thiazolidinediones increase insulin sensitivity acting mainly in peripheral tissues like adipose tissue, muscle, and less in the liver, increasing glucose utilization and decreasing glucose production. The mechanism by which the thiazolidinediones exert their effect is not fully understood. They bind to and activate one or more peroxisome proliferator-activated receptors (PPARs), which regulate gene expression in response to ligand binding (Vidal-Puig et al. 1997).
PPAR-gamma (one of the PPARs receptor family) was found predominantly in adipose tissue, pancreatic beta-cells, vascular endothelium, macrophages, and the central nervous system. On the other hand, PPAR-alpha is expressed mostly in liver, heart, skeletal muscle, and vascular walls. The various thiazolidinediones have differential effects on PPAR-gamma and PPAR-alpha receptors. Troglitazone and rosiglitazone are purely PPAR-gamma agonists, while pioglitazone also exerts some PPAR-alpha effects. This may account for the different effects that pioglitazone and rosiglitazone have on lipids.
Thiazolidinediones improve insulin action in skeletal muscle by facilitating glucose transport activity, thereby increasing the rates of muscle glycogen synthesis and glucose oxidation.
The insulin-sensitizing effect of thiazolidinediones may be related in part to the regulation of adipose tissue production of adipokines via PPAR-gamma activation
PPAR-gamma activation mediates weight gain by promoting increased feeding. This finding accounts, in part, for the weight gain associated with treatment (Lu et al. 2011; Ryan et al. 2011). Thiazolidinediones increase peripheral fat content (with a slight increase of weight) but reduce visceral fat. An interesting preserving pancreatic beta-cell function has been demonstrated.
Finally, new “dual” PPAR agonists (gamma and alpha) are being investigated to probe if they could effectively treat both hyperglycemia and hyperlipidemia.
10.2.4.2 Doses, Associations, and Efficacy
When used as monotherapy, they reduce hemoglobin HbA1c percentage between 0.8 and 1.5 %, showing the same potency as that of metformin
Thiazolidinediones could be administered as monotherapy or in combination with complementary antidiabetic drugs such as metformin, sulfonylureas, meglitinides, incretin mimetics, and DPP4 inhibitors. The association with insulin is not recommended and the combination with sulfonylurea has to be avoided in obese type 2 diabetic patients. The association of thiazolidinedione and metformin is very rational and very effective because metformin inhibits hepatic glucose production (improving insulin sensibility in the liver), whereas thiazolidinediones act by improving peripheral uptake and utilization of glucose in muscle and fat. In this way both reduce in a complementary action central and peripheral insulin resistance (Raskin et al. 2001; Kipnes et al. 2001).
10.2.4.3 Side Effects and Contraindications
The main side effects of thiazolidinediones are as follows: (1) potential liver toxicity although there are many trials showing improvement in patients with hepatosteatosis; (2) weight gain; (3) fluid retention and possible induction or worsening of heart failure in susceptible patients; and (4) bone fractures.
Weight gain: It is both dose-dependent and time-dependent and can be substantial (Fonseca et al. 2000). The most relevant clinical aspects of these drugs are summarized in Table 10.7
Table 10.7
Clinically relevant metabolic effects of thiazolidinediones
Glycemic effects |
Increased uptake and insulin-mediated utilization of glucose |
Glucose-lowering action without hypoglycemia |
Non-glycemic effects |
Lipids |
Conversion of small, dense LDL particles in large and sparse |
Increase of HDL cholesterol |
Lower triglycerides if they are increased (>200 mg/dl) |
Increase adipogenesis |
Decrease of free fatty acids in plasma |
Vascular |
Ameliorate endothelial dysfunction? |
Improve vascular resistance reducing a mean of 4 mm Hg in systolic and diastolic blood pressures |
Ameliorate the procoagulant state decreasing the PAI-1 and fibrinogen concentrations |
Decrease inflammation process measured by CRP |
Decrease the thickness of the carotid intima-media |
Reduce albuminuria |
Pancreatic beta cells |
Reduces the rate of loss of secretory function in animals |
Contraindications of TZDs are Type 1 diabetes, liver disorders, and pregnancy, and the relative contraindications are Obese Type 2 diabetic patients
10.2.5 Alpha-glucosidase Inhibitors
Two oral antidiabetic drugs, alpha-glucosidase inhibitors and lipase inhibitors, lower blood glucose modifying the intestinal absorption of carbohydrates and fat, respectively .
The alpha-glucosidase inhibitors, acarbose, miglitol, and voglibose, have been studied extensively in Europe and Japan; two of them, acarbose and miglitol, are available in the United States. They inhibit the upper gastrointestinal enzymes (alpha-glucosidases) that convert complex polysaccharide carbohydrates into monosaccharide in a dose-dependent fashion slowing the absorption of glucose. This action decreased postprandial blood glucose concentrations. Acarbose has a potential beneficial effect in postprandial hyperglycemia in both Type 1 and Type 2 diabetes patients. In older patients with type 2 diabetes, acarbose may also increase insulin sensitivity (Meneilly et al. 2000). These drugs also modify the secretion of gastrointestinal incretins.
10.2.5.1 Mechanisms of Action
Inhibitors competitively block alpha-glucosidase located in the brush border of the small intestine cells, enzymes for hydrolyzing essentials disaccharides, oligosaccharides, and polysaccharides to monosaccharides. Under normal circumstances, the carbohydrates are absorbed quickly in the first portion of the small intestine. Therefore, these drugs delay the absorption of carbohydrates along the entire small intestine.
10.2.5.2 Doses, Associations, and Efficacy
Efficacy—Alpha-glucosidase inhibitors are less potent than most of the antidiabetic drugs lowering HbA1c by 0.4–0.9 % (Van de Laar et al. 2005).
Dosing—Acarbose is available as 50 and 100 mg tablets, which should be taken with the first bite of each meal beginning with 50 mg three times daily. Flatulence, diarrhea, and abdominal discomfort are dose related and will almost always resolve if the dose is decreased. Few people tolerate more than 300 mg daily. One of the main advantages is that they do not produce hypoglycemia.
10.2.5.3 Side Effects and Contraindications
Although alpha-glucosidase inhibitors have been studied as monotherapy for the initial treatment of diabetes and even prediabetes, most of the guidelines do not consider using them as a first-line therapy because of reduced efficacy, expense, and poor tolerance.
The formal contraindications to treatment with the inhibitors of alpha-glucosidase are intestinal malabsorption syndromes, inflammatory bowel disease, intestinal obstruction, and liver failure. They are also contraindicated in severe renal failure, pregnancy and lactation, as well as in children less than 12 years.
10.2.6 GLP-1 Receptor Antagonist
10.2.6.1 Mechanism of Action
The two main incretin hormones, GIP and GLP-1, are released predominantly from L and K cells (in the proximal and distal intestine) following nutrient ingestion promoting insulin secretion, glucagon release inhibition, delaying gastric emptying, and enhancing satiety (resulting in weight control) with GLP-1 being much more effective than GIP (Van Genugten et al. 2013).
Incretin mimetics or enhancers are one of the latest groups of drugs available for the treatment of T2DM—incretin therapy. Given the novel role of incretin therapy in terms of reducing postprandial hyperglycemia, and the favorable effect on weight with the reduced incidence of hypoglycemia, the used of them as alternative options in T2DM was explored [28].
Some evidence alludes to incretins potentially increasing beta cell mass and altering disease progression, so it might be helpful to introduce these agents earlier in the treatment algorithm.
Incretin acts primarily by increasing the physiological effects mediated via the hormone GLP-1, which is secreted along with GIP by intestinal cells when food is ingested, probably via the neural and endocrine signals associated with feeding. GLP-1 and GIP have multiple actions that enhance beta-cell response in a glucose-dependent fashion. In T2DM, the incretin response is diminished (Nauck et al. 1986).
Two strategies can restore the GLP-1 signal: (1) inhibiting the ubiquitous enzyme dipeptidyl peptidase-4 (DPP-4), which rapidly degrades GLP-1 in vivo resulting in increased concentrations of endogenous GLP-1 called incretin enhancers or (2) using parenteral administration of GLP-1 mimetics or GLP-1 RAs (GLP-1 Receptor Agonists) that are DPP4 resistant (Druker 2006).
10.2.6.2 GLP-1 Mimetics
Currently four GLP-1 RAs are available.
The insulinotropic and extra-pancreatic effects of GLP-1 RAs have been described. In relation to glucose metabolism, GLP-1 increases insulin secretion and suppresses glucagon secretion; both mechanisms manifest only in the setting of hyperglycemia. In addition, GLP-1 induces satiety and has an effect on weight (Table 10.8) (Owens et al. 2013).
Table 10.8
Effects of incretin therapy in the treatment of T2DM
(a) Glucose-dependent stimulation of insulin secretion |
(b) Glucose-dependent suppression of glucagon secretion |
(c) Reduced gastrointestinal motility |
(d) Increased satiety |
(e) Increased beta-cell mass with inhibition of beta-cell apoptosis |
(f) Improvement of beta-cell function |
10.2.6.3 Pharmacological Profile of GLP-1
Pharmacology of GLP-1 can be appreciated in Table 10.9.
Table 10.9
Classification of GLP-1 RA according to pharmacological profile
Prandial GLP-1 RA | Exenatide |
Lixisenatide | |
Non-prandial GLP-1 RA | Extended release exenatide |
Liraglutide | |
Albiglutide | |
Semaglutide |
1. Prandial GLP-1 (Exenatide and Lixisenatide)
Exenatide is a synthetic form of exendin-4, a 39 amino-acid peptide isolate from the salivary secretions of the Gila monster with a 50 % homology with GLP-1 and is a potent agonist of the human GLP-1 receptor. Exenatide has a half-life of 2 h, so two injections daily are necessary. Different studies demonstrated that exenatide in combination with metformin, a DPP4 inhibitor, or rosiglitazone decreased post-prandial glycemia, and the most significant decrease was with rosiglitazone. When it was compared with glimepiride or glibenclamide, the decrease was almost the same. Another study compared exenatide with insulin glargine or biphasic insulin aspart, HbA1c was lower by both insulins, fasting plasma glycemia was lower with insulin glargine, and post-prandial glycemia improved with exenatide plus metformin and TZD (DeFronzo et al. 2008, 2010; Derosa et al. 2010, 2011) (Table 10.10).
Table 10.10
Comparison of pharmacological profile of GLP-1
Prandial | Non-prandial | |
|---|---|---|
GLP-1 RA | EXENATIDE LIXISENATIDE | LIRAGLUTIDE EXENATIDE—LAR |
Half-life | 2–5 h | 12 h-many days |
Effect on fasting glucose | Modest reduction | High reduction |
Effect on post-prandial glucose | High reduction | Modest reduction |
Gastric emptying | High impact | No effect |
Lixisenatide is a 44 amino-acid exendin-4 analog with an extended c-terminus. The half time of lixisenatide is 2.8 h, but the binding affinity for the GLP-1 receptor is fourfold greater than native GLP-1 that allows once daily injection. The Phase III GETGOAL program comprised 11 randomized studies that evaluated the efficacy and tolerability of lixisenatide 20 μg daily prandial in individuals with T2DM. Lixisenatide produces significant improvements in HbA1c, FPG, and PPG compared with insulin glargine or oral agents (Ahren et al. 2013; Riddle et al. 2013; Bolli et al. 2014; Fonseca et al. 2012; Pinget et al. 2013; Ratner et al. 2011; Lorenz et al. 2013; Kapitza et al. 2013).
2. Non-prandial GLP-1: Extended Release Exenatide, Liraglutide, Albiglutide, and Semaglutide
These are molecules that rely on different mechanisms to delay their absorption from the subcutaneous tissue and extend their duration of action (Table 10.10).
2a
 Stem Cells Derived Insulin-Secreting Cells for Insulin-Dependent Diabetes Mellitus: Exploiting Laboratory Discoveries
Peripheral Signals and Food Intake Control
Stem Cells Derived Insulin-Secreting Cells for Insulin-Dependent Diabetes Mellitus: Exploiting Laboratory Discoveries
Peripheral Signals and Food Intake Control
 Ileal Transposition Surgery: Mechanisms of Weight Loss and Diabetes Improvements
Ileal Transposition Surgery: Mechanisms of Weight Loss and Diabetes Improvements
 Left Gastric Artery Embolization to Treat Obesity: Rationale, Technique, Experimental, and Clinical Studies
Left Gastric Artery Embolization to Treat Obesity: Rationale, Technique, Experimental, and Clinical Studies
 Endoscopic Therapeutic Options for Type 2 Diabetes
Endoscopic Therapeutic Options for Type 2 Diabetes
 Sleeve Gastrectomy and Transit Bipartition
Sleeve Gastrectomy and Transit Bipartition




Extended release exenatide is a formulation that encapsulates microspheres made of biodegradable polymer that allows once weekly administration. The Phase III clinical development programme of exenatide once weekly involved a series of trials known as the DURATION trials. Safety and efficacy of Exenatide once weekly were compared with exenatide twice daily, pioglitazone or sitagliptin, metformin, liraglutide, and insulin glargine (DURATION 1–6), respectively (Bergenstal et al. 2010; Russell-Jones et al. 2012; Buse et al. 2012; Diamant et al. 2010; Raskin et al. 2005; Blevins et al. 2011). Extended release exenatide was superior in lowering PPG, had a low incidence of hypoglycemia but a greater incidence of gastrointestinal adverse events. The sustained elevated plasma concentrations of exenatide once weekly formulation induced tachyphylaxis with a progressive inhibition of gastric emptying, a key factor related with the reduction of PPG (Nauck et al. 2011).
Related posts:
Stem Cells Derived Insulin-Secreting Cells for Insulin-Dependent Diabetes Mellitus: Exploiting Laboratory Discoveries
Peripheral Signals and Food Intake Control
Ileal Transposition Surgery: Mechanisms of Weight Loss and Diabetes Improvements
Left Gastric Artery Embolization to Treat Obesity: Rationale, Technique, Experimental, and Clinical Studies
Endoscopic Therapeutic Options for Type 2 Diabetes
Sleeve Gastrectomy and Transit Bipartition
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
